Running the duplicate reads removal
The tool is found in the Toolbox:
Toolbox | Resequencing Analysis (![]() ) | Remove Duplicate Mapped Reads (
) | Remove Duplicate Mapped Reads (![]() )
)
This opens a dialog where you can select mapping results in read tracks (![]() ) format. Clicking Next allows you to set the threshold parameters as displayed in figure 21.37.
) format. Clicking Next allows you to set the threshold parameters as displayed in figure 21.37.
Figure 21.37: Setting the stringency for merging similar reads.
The parameter is explained in detail in Algorithm details and parameters.
Clicking Next will reveal the output options. The main output is a list of the reads that remain after the duplicates have been removed. In addition, you can get the following output:
- List of duplicate sequences
- These are the sequences that have been removed.
- Report
- This is a brief summary report with the number of reads that have been removed (see an example in figure 21.38).
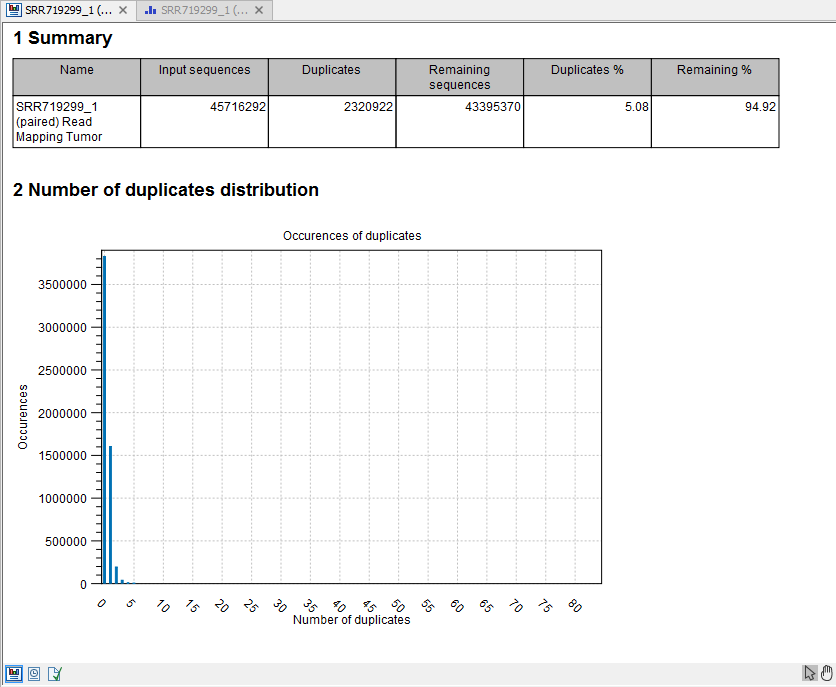
Figure 21.38: Summary statistics on the duplicate mapped reads.