Remove Germline Variants
Running the variant caller on a case and control sample separately and filtering away variants found in the control data set does not always give a satisfactory result as many variants in the control sample have not been called. This is often due to lack of read coverage in the corresponding regions or too stringent parameter settings. Therefore, instead of calling variants in the control sample, the Remove Germline Variants tool can be used to remove variants found in both samples from the set of candidate variants identified in the case sample.
Toolbox | Remove
Variants (![]() ) |
Remove Germline Variants
) |
Remove Germline Variants
The variant track from the case sample must be used as input. When clicking Next, you are asked to supply the number of reads in the control data set that should support the variant allele in order to include it as a match (see figure 23.3). All the variants where at least this number of control reads show the particular allele will be filtered away in the result track.
Please note that variants, which have no coverage in the mapped control reads will be reported too. You can identify them by looking for a 0 value in the column 'Control coverage'.
The following annotations will be added to each variant not found in the control data set:
- Control count
- For each allele the number of reads supporting the allele.
- Control coverage
- Read coverage in the control dataset for the position in which the allele has been identified in the case dataset.
- Control frequency
- Percentage of reads supporting the allele in the control sample.
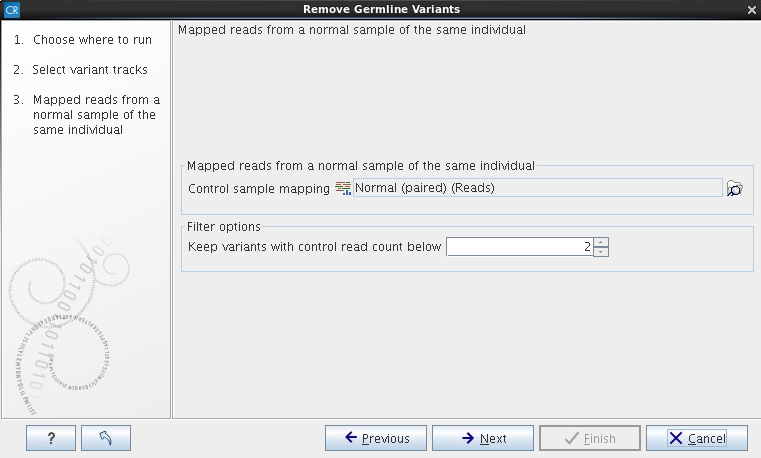
Figure 23.3: Specify here the read mapping of the control sample
and the minimum number of reads, which should include the variant before it
will be removed as germline variant.
The filter option can be used to set a threshold for which variants should be kept. In the dialog shown in figure 23.3 the threshold is set at two. This means that if a variant is found in only two or less of the control reads, it will be filtered away.