Filter Somatic Variants (WES)
If you are analyzing a list of variants that have been detected in a tumor or blood sample where no control sample is available from the same patient, you can use the "Filter Somatic Variants (WES)" ready-to-use workflow to identify potential somatic variants. The purpose of this ready-to-use workflow is to use publicly available (or your own) databases, with common variants in a population, to extract potential somatic variants whenever no control/normal sample from the same patient is available.
The "Filter Somatic Variants (WES)" ready-to-use workflow accepts variant tracks (![]() ) (e.g. the output from the Identify Variants ready-to-use workflow) as input. In cases with heterozygous variants, the reference allele is first filtered away, then variants outside the targeted region are removed, and lastly, variants found in the Common dbSNP, 1000 Genomes Project, and HapMap databases are deleted. Variants in those databases are assumed to not contain relevant somatic variants.
) (e.g. the output from the Identify Variants ready-to-use workflow) as input. In cases with heterozygous variants, the reference allele is first filtered away, then variants outside the targeted region are removed, and lastly, variants found in the Common dbSNP, 1000 Genomes Project, and HapMap databases are deleted. Variants in those databases are assumed to not contain relevant somatic variants.
Please note that this tool will likely also remove inherited cancer variants that are present at a low percentage in a population.
Next, the remaining somatic variants are annotated with gene names, amino acid changes, conservation scores and information from COSMIC (database with known variants in cancer), ClinVar (known variants with medical impact) and dbSNP (all known variants).
To run the Filter Somatic Variants tool, go to:
Toolbox | Ready-to-Use Workflows | Whole Exome Sequencing (![]() ) | Filter Somatic Variants (
) | Filter Somatic Variants (![]() )
)
- Double-click on the Filter Somatic Variants tool to start the analysis. If you are connected to a server, you will first be asked where you would like to run the analysis. Next, you will be asked to select the variant track you would like to use for filtering somatic variants. The panel in the left side of the wizard shows the kind of input that should be provided (figure 14.16). Select by double-clicking on the reads file name or clicking once on the file and then clicking on the arrow pointing to the right side in the middle of the wizard.
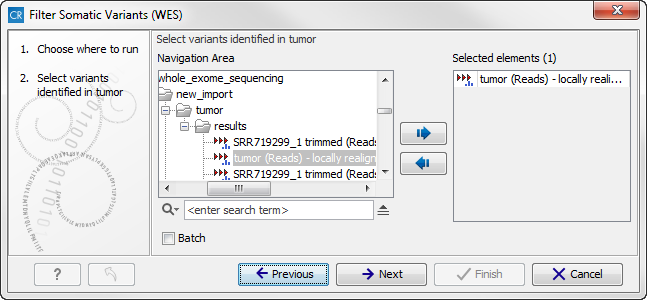
Figure 14.16: Select the variant track from which you would like to filter somatic variants.Click on the button labeled Next.
- In the next step you will be asked to specify which of the 1000 Genomes populations that should be used for annotation (figure 14.17).
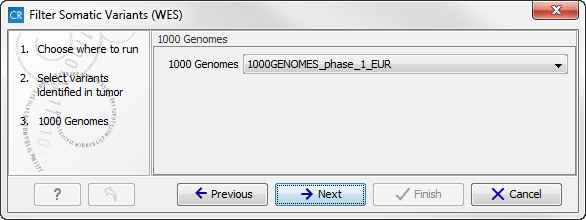
Figure 14.17: Specify which 1000 Genomes population to use for annotation.Click on the button labeled Next.
- In this wizard step, you are asked to supply a track containing the targeted regions (figure 14.18). Select the track by clicking on the folder icon (
 ) in the wizard.
) in the wizard.
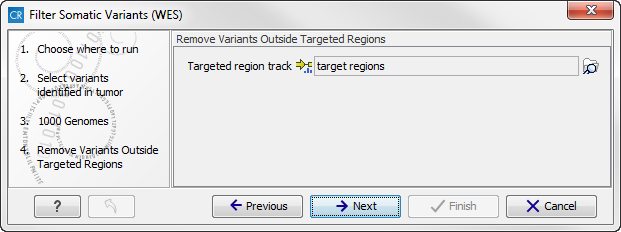
Figure 14.18: Select your target regions track.Click on the button labeled Next.
- The next wizard step will once again allow you to specify the 1000 Genomes population that should be used, this time for filtering out variants found in the 1000 Genomes project (figure 14.19).
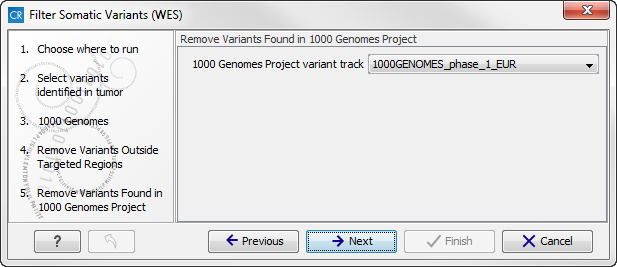
Figure 14.19: Specify which 1000 Genomes population to use for filtering out known variants.Click on the button labeled Next.
- The next wizard step (figure 14.20) concerns removal of variants found in the HapMap database. Select the population you would like to use from the drop-down list. Please note that the populations available from the drop-down list can be specified with the Data Management (
 ) function found in the top right corner of the Workbench (see Download and configure reference data).
) function found in the top right corner of the Workbench (see Download and configure reference data).
Figure 14.20: Specify which HapMap population to use for filtering out known variants. - Click on the button labeled Next to go to the last wizard step (shown in figure 14.21).
Figure 14.21: Check the selected parametes by pressing "Preview All Parameters".Pressing the button Preview All Parameters allows you to preview all parameters. At this step you can only view the parameters, it is not possible to make any changes. Choose to save the results and click on the button labeled Finish.
Two types of output are generated:
- Somatic Candidate Variants Track that holds the variant data. This track is also included in the Genome Browser View. If you hold down the Ctrl key (Cmd on Mac) while clicking on the table icon in the lower left side of the View Area, you can open the table view in split view. The table and the variant track are linked together, and when you click on a row in the table, the track view will automatically bring this position into focus.
- Genome Browser View Filter Somatic Variants A collection of tracks presented together. Shows the somatic candidate variants together with the human reference sequence, genes, transcripts, coding regions, and variants detected in ClinVar, COSMIC, 1000 Genomes, and the PhastCons conservation scores (see figure 14.22).
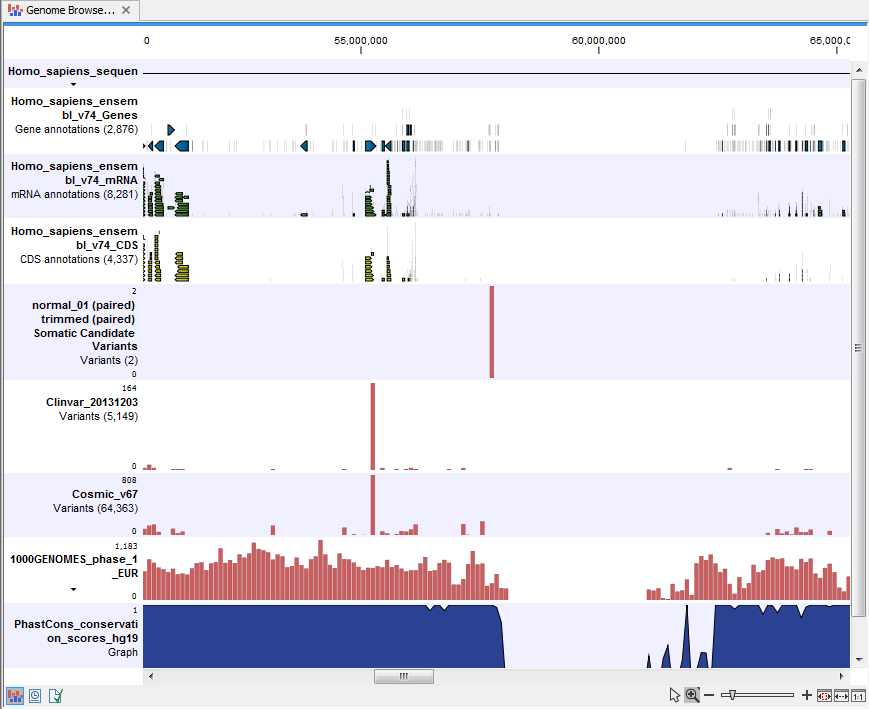
Figure 14.22: The Genome Browser View showing the annotated somatic variants together with a range of other tracks.
To see the level of nucleotide conservation (from a multiple alignment with many vertebrates) in the region around each variant, a track with conservation scores is added as well. Mapped sequencing reads as well as other tracks can be easily added to this Genome Browser View. By double clicking on the annotated variant track in the Genome Browser View, a table will be shown that includes all variants and the added information/annotations (see figure 14.23).
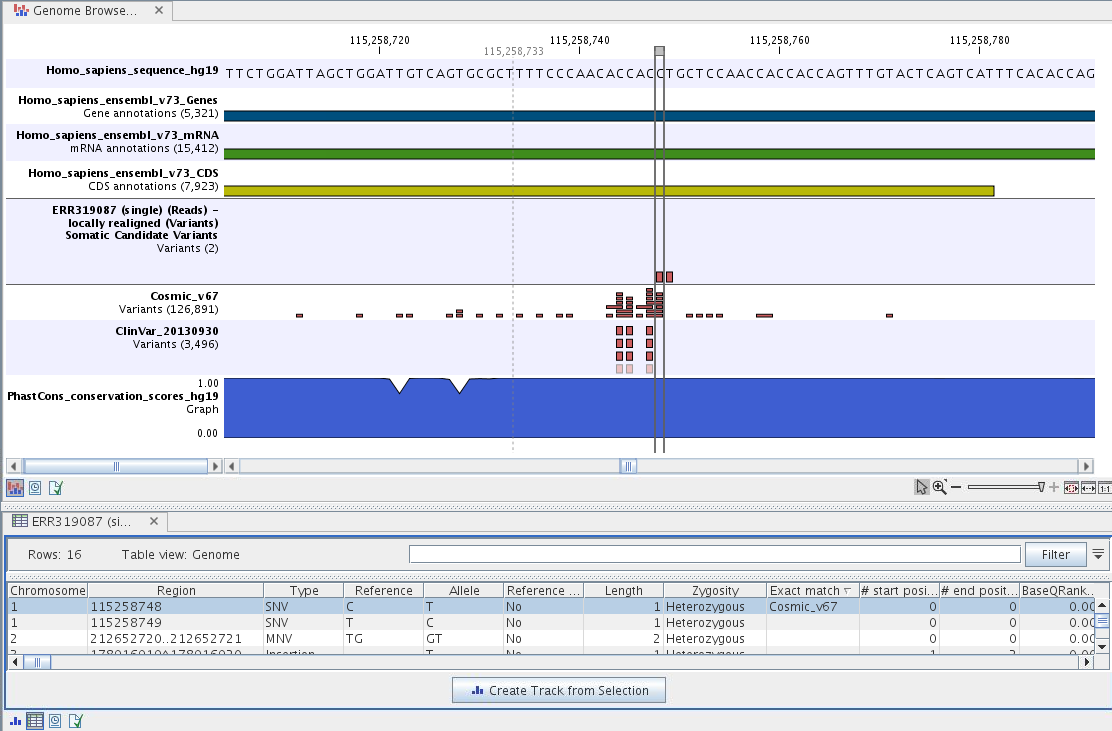
Figure 14.23: The Genome Browser View showing the annotated somatic variants together with a range of other tracks.
Adding information from other sources may help you identify interesting candidate variants for further research. E.g. known cancer associated variants (present in the COSMIC database) or variants known to play a role in drug response or other clinical relevant phenotypes (present in the ClinVar database) can easily be identified. Further, variants not found in the COSMIC and/or ClinVar databases, can be prioritized based on amino acid changes in case the variant causes changes on the amino acid level.
A high conservation level, between different vertebrates or mammals, in the region containing the variant, can also be used to give a hint about whether a given variant is found in a region with an important functional role. If you would like to use the conservation scores to identify interesting variants, we recommend that variants with a conservation score of more than 0.9 (PhastCons score) is prioritized over variants with lower conservation scores.
It is possible to filter variants based on their annotations. This type of filtering can be facilitated using the table filter found at the top part of the table. If you are performing multiple experiments where you would like to use the exact same filter criteria, you can create a filter that can be saved and reused. To do this:
Toolbox | Identify Candidate Variants (![]() ) | Create Filter Criteria (
) | Create Filter Criteria (![]() )
)
This tool can be used to specify the filter and the Annotate Variants workflow should be extended by the Identify Candidate Tool (configured with the Filter Criterion). The CLC Cancer Research Workbench reference manual has a chapter that describes this in detail (http://clccancer.com/software/#downloads, see chapter: "Workflows" for more information on how pre-installed workflows can be extended and/or edited).
Note! Sometimes the databases (e.g. COSMIC) are updated with a newer version, or maybe you have your own version of the database. In such cases you may wish to change one of the used databases. This can be done with "Data Management" function, which is described in Download and configure reference data.