Running the Extract Differentially Expressed Genes tool
After you start the tool, you are presented with a wizard where you can choose the experiment that you would like to create a track of. The Extract Differentially Expressed Genes tool can be run on two types of experiments:
- Experiments with associated genomic information, such as those created using expression tracks from the RNA-Seq Analysis tool.
- Experiments without associated genomic information, such as those created using samples from the legacy RNA-Seq Analysis tool.
In the case where the experiment has associated genomic information, the Extract Differentially Expressed Genes tool will automatically infer these and the wizard will jump directly to the filtering step, as shown in figure 28.56.
In the case where the experiment does not have associated genomic information, you will first need to specify how the genomic information should be obtained in the Parameters step of the Extract Differentially Expressed Genes tool (figure 28.57).
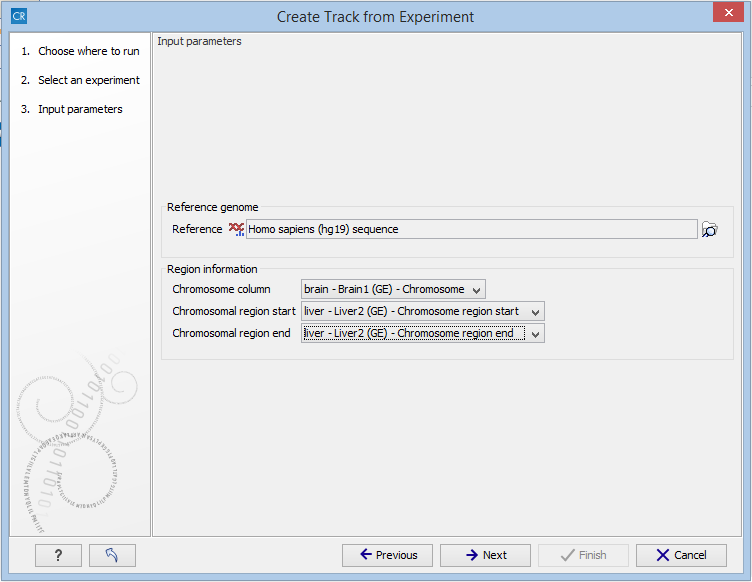
Figure 28.55: The "Input parameters" step in the Extract Differentially Expressed Genes tool.
In the Input parameters step, you must specify the following parameters:
- Reference genome. The chosen genome will be used as the reference genome for the resulting track.
- Chromosome column. The column containing the chromosome names must be chosen from the drop-down menu.
- Chromosomal region start. The column containing the start of the genomic regions must be chosen from the drop-down menu.
- Chromosomal region end. The column containing the end of the genomic regions must be chosen from the drop-down menu.
Note! The drop-down menus will only contain the columns that potentially represent the information required by the given parameter. If the experiment does not contain any columns that potentially represent the required genomic information, the drop-down menus may appear empty. In this case, it is not possible to convert the given experiment to a track.
In the Filtering step (figure 28.56), you have the following options:
- Filter based on statistical analysis results This allows to filter which annotations are transferred to the track on the basis of the statistical analysis. To enable filtering, check the Filter based on statistical analysis results checkbox. The filtering option is only available if a statistical analysis has previously been carried out on the Experiment, and the drop-down menu will only contain the statistical analyses that are present on the Experiment.
- Statistical analysis Allows you to choose statistical analysis from the drop-down list. The selection of available statistical analyses depends on which tests have been used when you set up the experiment that you are about to convert to track format.
- Type of p-value This drop-down menu allows you to select between raw and corrected p-values (see Corrected p-values). Only the types of p-values available for the given statistical analysis will be present in the drop-down menu.
- Maximum p-value In this input field, you can enter the maximum allowed p-value, as a number between 0 and 1. If you do not want any filtering based on p-value, enter 1.
- Minimum fold-change value You can also specify the minimum allowed fold-change value as a number greater than zero. If you do not want any filtering based on fold-change, enter 0.
The fold change values are stored as different columns in the experiment, depending on the statistical analysis performed. The Extract Differentially Expressed Genes tool will automatically use the fold-change column appropriate for the different statistical analyses:
- Kal's Z-test (see Kal et al.'s test (Z-test)): Proportions fold change.
- Baggerley's test(see Baggerley et al.'s test (Beta-binomial)): Weighted proportions fold change.
- T-test (see T-tests): Fold change.
- ANOVA (see ANOVA): Max fold change.
- Empirical analysis of DGE (see Empirical analysis of DGE): Fold change.
If the chosen statistical analysis was performed on several pairs of groups, there will be an output track for each tested pair of groups. For example, if the same statistical analysis has been carried out on 'group 1 vs. group 2' and 'group 1 vs. group 3', then the output will contain two tracks, where one is filtered according to the 'group 1 vs. group 2' analysis results and the other one is filtered according to the 'group 1 vs. group 3' analysis results.
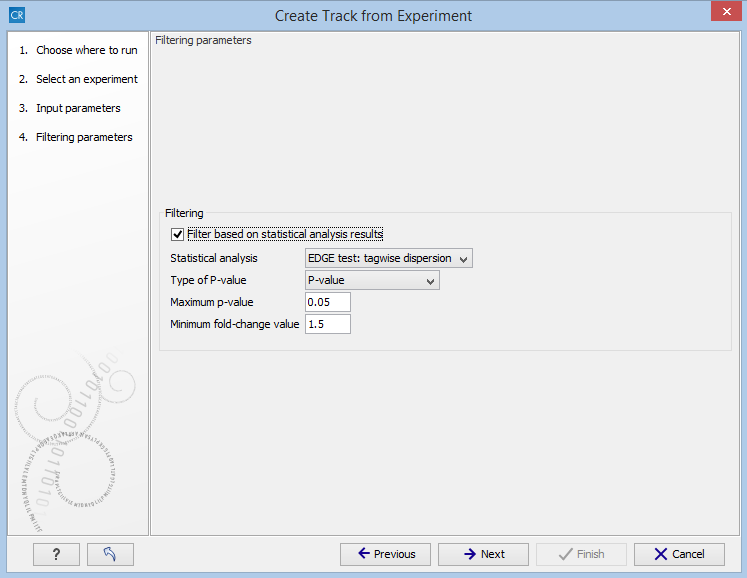
Figure 28.56: The filtering step in the Extract Differentially Expressed Genes tool.
Figure 28.57: The result handling step in the Extract Differentially Expressed Genes tool.
When running the Extract Differentially Expressed Genes tool as part of a workflow, there are a few differences in how the parameters are set (see figure 28.58).
- The Source of genomic information parameter determines the behavior of the algorithm if the incoming experiment is not coupled to a genome. If the value of this parameter is set to Require genomic information in experiment, then the algorithm will expect the incoming experiment to be coupled to a genome, and will fail with an error alerting the user in case the experiment does not fulfill this criterion. If the value of the parameter is set to Automatic: use genomic information if available, then the algorithm will still use the genomic information in a genome-coupled experiment. But if this information is not available, the algorithm will attempt to use the information specified by the user in the workflow parameters. Note: If the incoming experiment is coupled to a genome (as will usually be the case), the value of this parameter makes no difference.
- In a workflow setting, the column titles for the chromosome, region end and region start fields can be specified as texts. These fields may be left empty, if the incoming experiment contains the genomic information. If filling out these fields, note that the format for this text is very strict, and must exactly match the text appearing in the drop-down menu when running the tool from the toolbox. For example, if 'Chromosome' is a sample-specific column, for a sample called 'Liver (GE)' in the 'liver' group in the experiment, then the column name text will be: 'liver - Liver (GE) - Chromosome'.
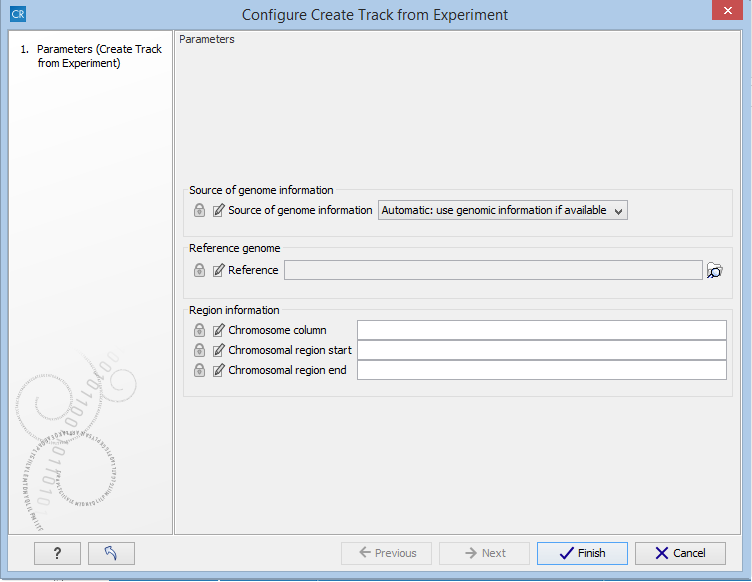
Figure 28.58: Setting the parameters for the Extract Differentially Expressed Genes tool in a workflow