Probabilistic variant detection
The purpose of the Probabilistic Variant Caller is to identify variants in a sample by using a probabilistic model built from read mapping data. This tool can detect variants in data sets from haploid (e.g. Bacteria), diploid (e.g. Human) and polyploid organisms (e.g. Cancer and higher plants) with a high sensitivity and specificity.
The algorithm used is a combination of a Bayesian model and a Maximum Likelihood approach to calculate prior and error probabilities for the Bayesian model.
Parameters are calculated on the mapped reads alone. The reference sequence is not considered at this stage. After observing a certain combination of nucleotides from the reads at every position in the genome, the probability for each combination of alleles is calculated. These probabilities are then used to determine which one of the allele combinations is the most likely combination for each position. In the case where the ploidy is expected to be 2, the types of cases considered would be homozygous A/A, heterozygous A/G, heterozygous A/C and so on. In the case where the ploidy is expected to be 3, the cases considered would be homozygous A/A/A, heterozygous A/G/C, heterozygous A/C/C and so on. This can then be compared with the reference allele to find out if it is different from the reference sequence and therefore can be called as a variant. Please refer to the white paper at http://www.clcbio.com/white-paper/ for more information including benchmarks.
Variants that are adjacent are reported as one. E.g. two SNVs next to each other will be reported as one MNV. Similarly, an SNV and an adjacent deletion will be reported as one replacement. Note that variants are only reported as one when they are supported by the same reads.
The size of insertions and deletions that can be found depend on how the reads are mapped: Only indels that are spanned by reads will be detected. This means that the reads have to align both before and after the indel. In order to detect larger insertions and deletions, please use the InDels and Structural Variation tool instead.
Please note that the variants reported by the structural variation tool can be fed into the local realignment tool to re-adjust the alignment of the reads to span the indels, making some of the indels detected by the structural variation ready to be picked up by the probabilistic variant detection.
Probabilistic Variant Detection is not designed to detect low frequency alleles. More specifically, it was not designed for calling variants that are not present in allelic frequencies that are in accordance with the ploidy assumption. Increasing the coverage will lead to higher confidence in the variants that are called, but will not result in the calling of low frequency variants. We recommend using the Low Frequency Variant Detection tool for this purpose (see Low Frequency Variant Detection) .
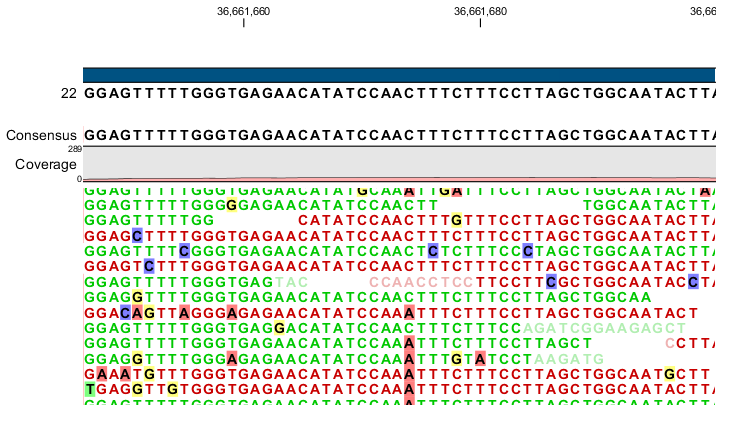
Figure 35.8: An example of a heterozygous variant surrounded by a lot of noise from sequencing errors.
Subsections
- Calculation of the prior and error probabilities
- Calculation of the likelihood
- Calculation of the posterior probability for each site type at each position in the genome
- Comparison with the reference sequence and identification of candidate variants
- Posterior filtering and reporting of variants
- Running the variant detection
- Setting ploidy and genetic code
- Reporting the variants found