Extract sequences from tracks
It is possible to extract sequences from tracks. The sequence of interest can be selected by dragging the mouse over the region of interest followed by a right click on the reads and a click on Extract sequences (figure 18.19).
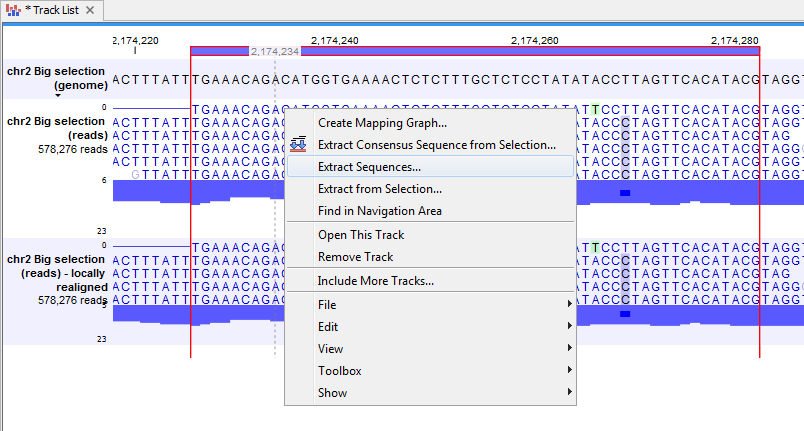
Figure 18.19: Extract sequences from tracks.
This opens up the dialog shown in figure 18.20 that allows specification of whether the selected sequences should be extracted as single sequences or as a list of sequences.
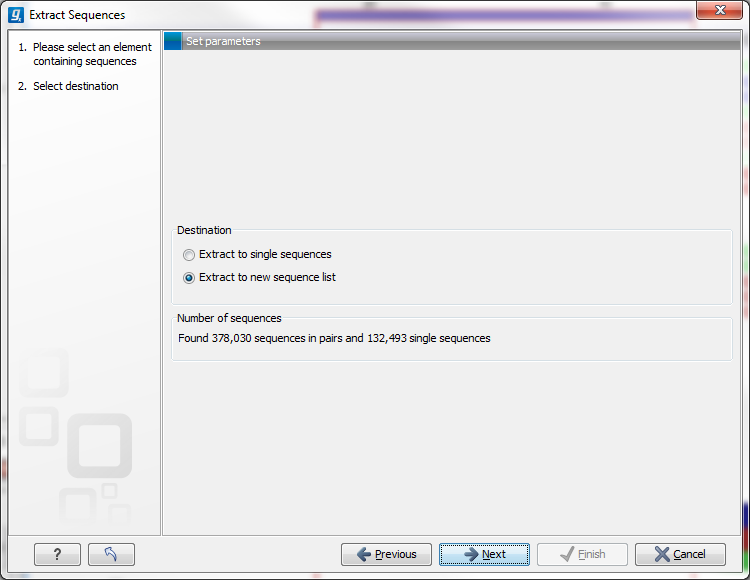
Figure 18.20: Select destination for extracted sequences.
Right clicking on the reads also enable the option Extract from selection, a function that corresponds to the Extract from selection described in section 31.7.5 although with small differences. Common for both versions of the Extract from selection function is that when extracting reads in an interval, only reads that are completely covered by the selection will be part of the extracted sequence, which in turn means that the tool can be used to extract only a subset of reads.
Clicking Extract from selection opens up the dialog shown in figure 18.21.
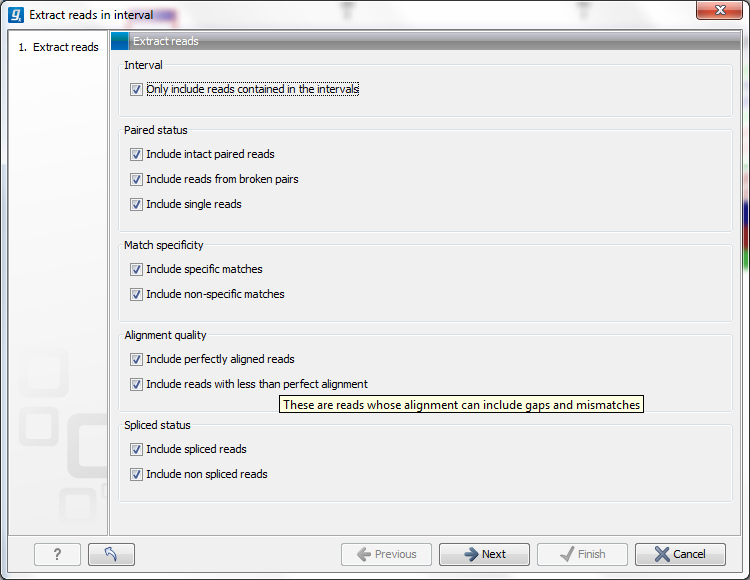
Figure 18.21: Select the reads to include.
The purpose of this dialog is to let you specify which kinds of reads you wish to include. Per default all reads are included.
The options are:
- Interval
- Only include reads contained within the intervals
- Only reads that are included within the selection will be extracted. Reads that continue outside the selected area are not included.
- Paired status
- Include intact paired reads
- When paired reads are placed within the paired distance specified, they will fall into this category. Per default, these reads are colored in blue.
- Include paired reads from broken pairs
- When a pair is broken, either because only one read in the pair matches, or because the distance or relative orientation is wrong, the reads are placed and colored as single reads, but you can still extract them by checking this box.
- Include single reads
- This will include reads that are marked as single reads (as opposed to paired reads). Note that paired reads that have been broken during assembly are not included in this category. Single reads that come from trimming paired sequence lists are included in this category.
- Match specificity
- Include specific matches
- Reads that only are mapped to one position.
- Include non-specific matches
- Reads that have multiple equally good alignments to the reference. These reads are colored yellow per default.
- Alignment quality
- Include perfectly aligned reads
- Reads where the full read is perfectly aligned to the reference sequence (or consensus sequence for de novo assemblies). Note that at the end of the contig, reads may extend beyond the contig (this is not visible unless you make a selection on the read and observe the position numbering in the status bar). Such reads are not considered perfectly aligned reads because they don't align in their entire length.
- Include reads with less than perfect alignment
- Reads with mismatches, insertions or deletions, or with unaligned nucleotides at the ends (the faded part of a read).
- Spliced status
- Include spliced reads
- Reads that are across an intron.
- Include non spliced reads
- Reads that are not across an intron.