Extending the Kraken2 external application for paired data
Here, we expand upon the configuration described in the Kraken2 example to create a Kraken2 external application for analyzing paired sequence data.
The steps involved in updating the original configuration, which handled single end data, involve:
- Updating the external application command, changing settings where needed and adding any new parameters necessary.
- Linking outputs from Kraken2 to an NGS importer that can correctly import pairs of files containing paired end sequences into sequence lists in the CLC software.
- Editing the included script to update the
Kraken2command so it handles paired data, and ensuring it handles all the inputs and outputs as needed.
Below, we describe these steps in the order listed, but the order they are carried out does not matter.
Updates to the external application command line and top level configuration
The command and configuration shown in figure 16.39 takes into account how Kraken2 handles paired sequence data and how CLC software handles paired sequence data.
Kraken2:
- Kraken2 expects paired data in 2 files, one containing the first member of each pair, and another containing the second member of each pair.
- Kraken2 returns paired sequence data in 2 files, one containing the first member of each pair, and another containing the second member of each pair.
CLC software:
- The FASTQ exporter can export paired sequence data to 2 files. (The FASTA importer, used in the single end example, cannot.)
- Each input parameter in the external application general command represents a single input that a CLC software user will be prompted for. Here, a sequence list selected by the user will be exported to 2 FASTQ format files, which are then passed to the included script. We must handle this situation, where a single parameter in the command is associated with more than one file.
- The Illumina high-throughput sequence importer can import pairs of files as paired sequence data. Information in the filenames is used to determine which file contains the first member of a pair and which contains the second member of a pair.
Using this knowledge, we can adjust the external application accordingly:
- The
Sequences to analyzeparameter is configured to use the FASTQ exporter, to export paired sequence data to 2 files (figure 16.38). - The
Sequences to analyzeparameter is used to pass 2 files to the included script, so that parameter is put in quotation marks in the Command line field (figure 16.39). - For each set of paired sequences generated by Kraken2, there are 2 parameters in the external application command line, one for each file (figure 16.39).
The parameter names for outputs from the third party tool are not important, as they are not seen by end users, however, for transparency, we reflected our assumption that the data provided will be in forward-reverse orientation in the parameter names, i.e.
Forward classified seqs,Reverse classified seqs,
Forward unclassified seqs, andReverse unclassified seqs. - For the sequence output parameters, we supply filenames that match our desired handling of the FASTQ files by the Illumina high-through sequencing importer: files with the first member of each pair have names ending with
_R1.fastq, files with the second member of each pair have names ending with_R2.fastq.For details of how filenames are interpreted, please refer to https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual= llumina.html.
- We link the parameters for
Kraken2sequence outputs with the Illumina high-throughput sequencing importer. Details of how this is done are provided below.
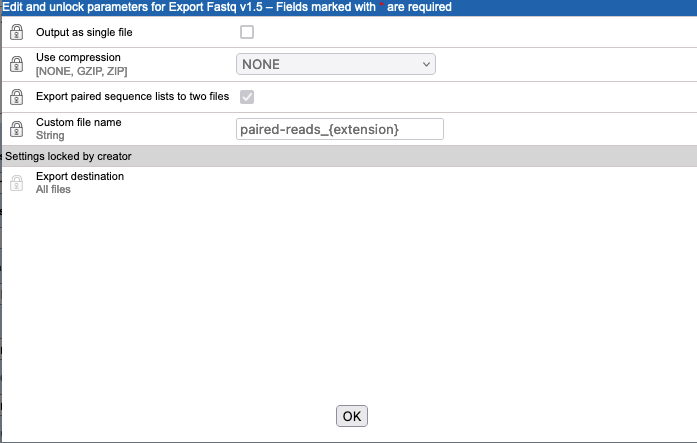
Figure 16.38: Clicking on the "Edit parameters" button for the "Sequences to analyze" parameter opens up a configuration window for the FASTQ exporter. The default settings are shown here, with the "Export paired sequence lists in two files" option enabled.
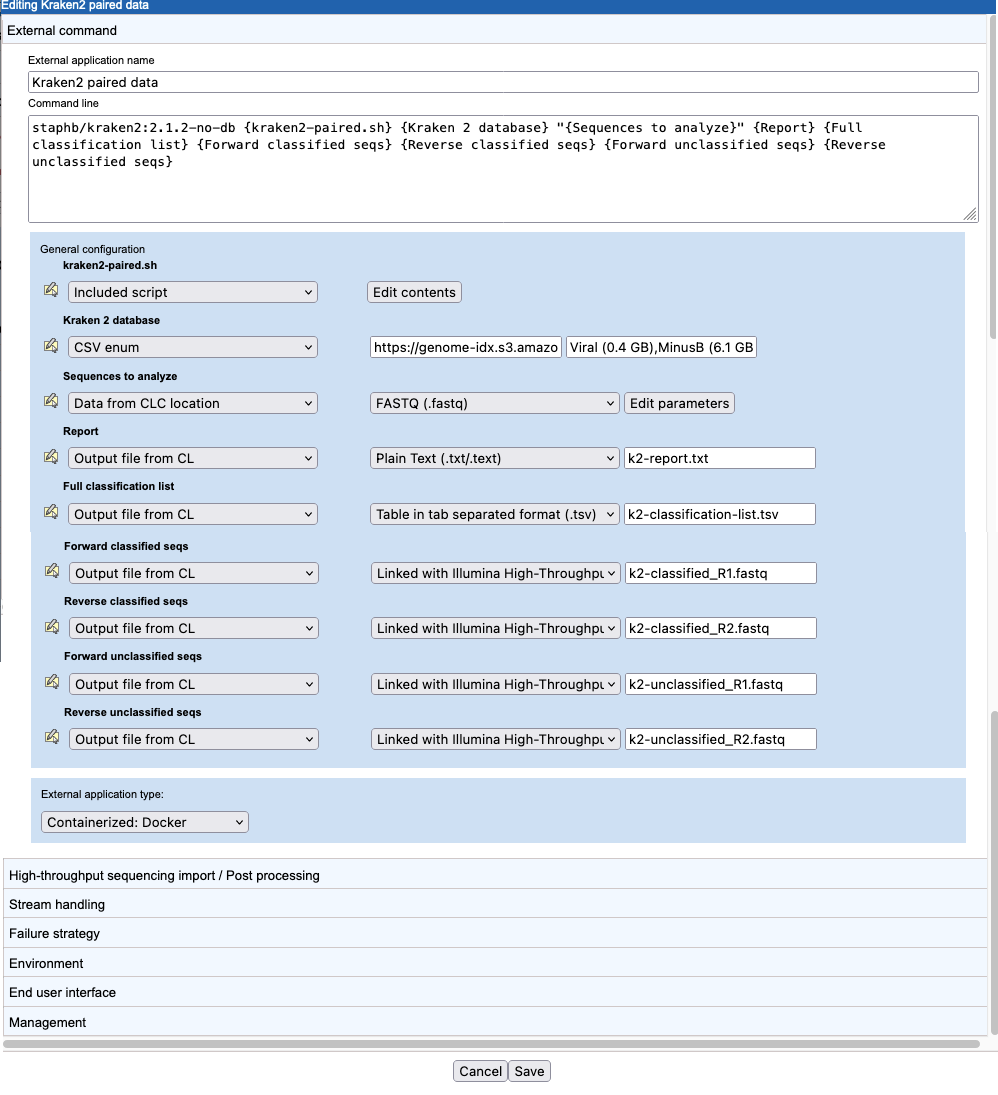
Figure 16.39: The external application command and top level configuration to support running Kraken2 with paired sequences as input and for handling the paired sequences Kraken2 generates as output.
Linking to a High-throughput importer
To handle the import of paired reads, a high-throughput sequence (NGS) importer is needed. Such importers require more configuration than the standard importers, i.e. those that can be directly selected in the "General configuration" area.
The steps to configure the import the 2 FASTQ files containing the classified sequences output by Kraken2 are described below. The same steps then need to be done to configure the import of the unclassified sequences.
- Use the default setting: "No standard import or map to high-throughput sequencing importer" for the
Forward classified seqsandReverse classified seqsparameters. - Click on the High-throughput sequencing import / Post-processing tab (below the general configuration area) to open it.
- Click on Add new and select an importer. Here, we select the Illumina importer (figure 16.40).
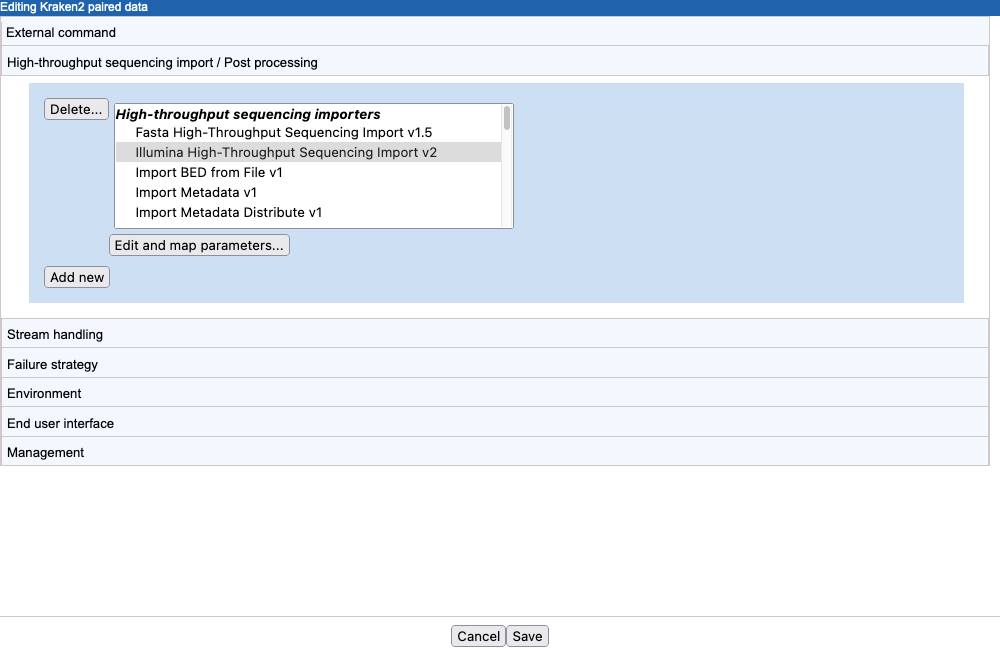
Figure 16.40: A new import/post processing element has been created, and the Illumina high-throughput sequencing importer has been selected. - Click on Edit and map parameters....
- Configure the importer.
Inputs All parameters in the Command line that are configured as type "Output file from CL" are available for selection as inputs to the importer. Select the 2 for the classified sequences output:
Forward classified seqsandReverse classified seqs(figure 16.41).Other importer settings Keep the default settings for all the other parameters.
- Click on the OK button to finish and save the importer configuration.
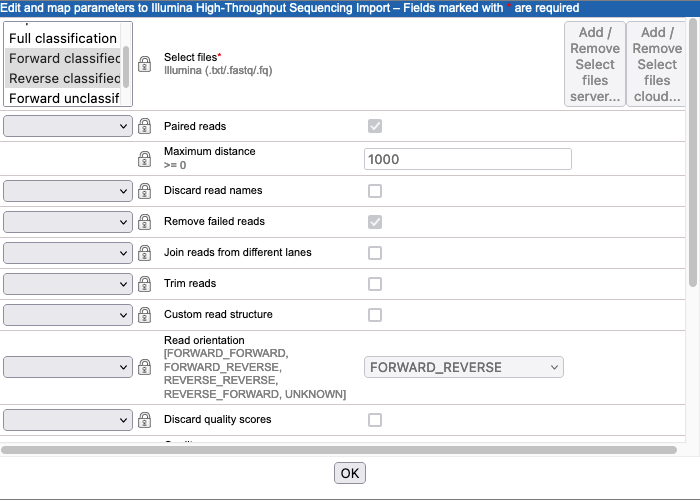
Figure 16.41: The Illumina High-Throughput Sequencing Import configuration. Here, the parameters corresponding to the 2 FASTQ files containing classified sequences have been selected. values.
The Forward classified seqs and Reverse classified seqs parameters in the "General configuration" area will be updated with the link to the Illumina importer (figure 16.42). If you don't see this immediately, save the external application configuration and re-open it.

Figure 16.42: The information in the "General configuration" area is updated to reflect the link between output parameters and the Illumina importer.
Updates to the included script
The included script must be updated: Kraken2 command needs to be adjusted and changes made to the external application configuration need to be handled.
Changes to the Kraken2 command:
--pairedhas been added to indicate that the sequences being provided are paired.k2-classified#.txtandk2-unclassified#.txtare given as values for the parameters--classified-outand--unclassified-outrespectively. The#symbol is a convention used byKraken2when working with paired data.
The Kraken2 command line is described at
https://github.com/DerrickWood/kraken2/wiki/Manual.
The other changes in the script are to handle the increased number of files being output by Kraken2 (figure 16.43).
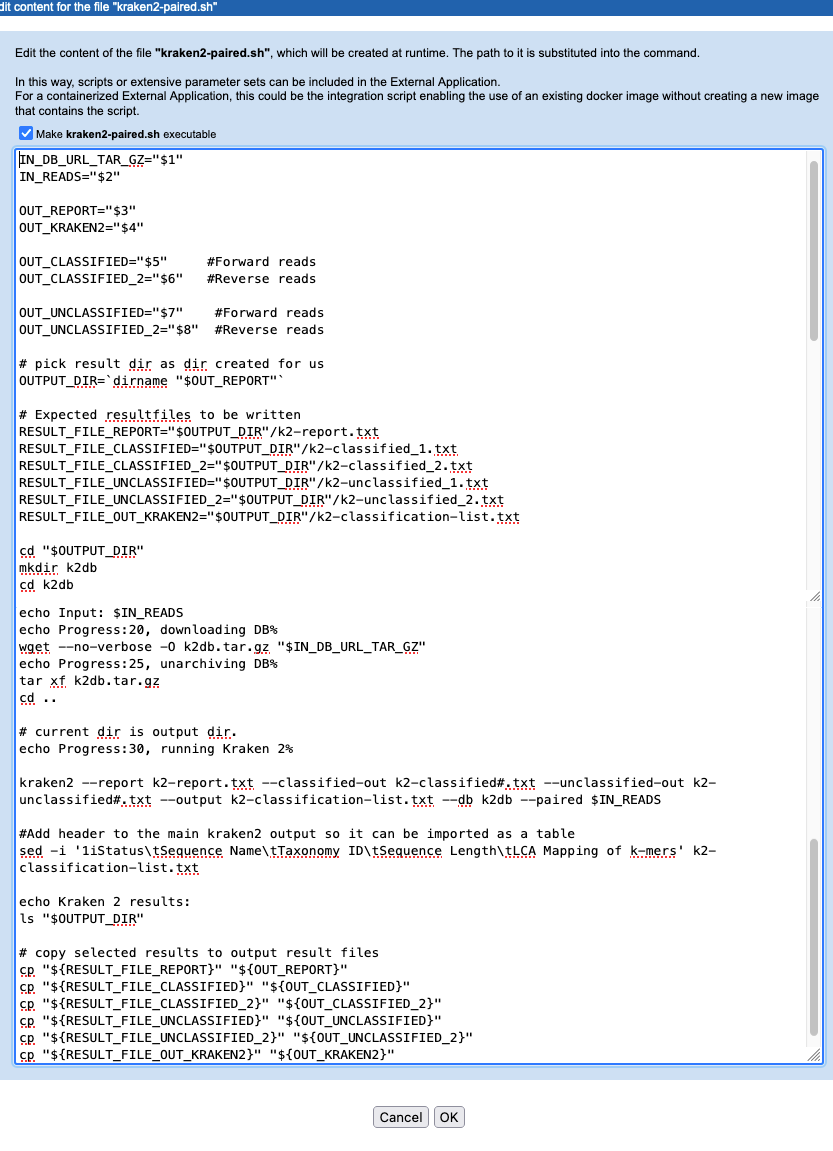
Figure 16.43: The kraken2-paired.sh script, with the Kraken2 command updated to indicated that the data to analyze is paired, and with variables added to handle paired sequence data being passed to the script and out of the script.