The Align Protein Structure algorithm
Any approach to structure alignment must make a trade-off between alignment length and alignment accuracy. For example, is it better to align 200 amino acids at an RMSD of 3.0 Å or 150 amino acids at an RMSD of 2.5 Å? The Align Protein Structure algorithm determines the answer to this question by taking the alignment with the higher TM-score. For an alignment focused on a protein of length
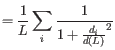
where ![]() runs over the aligned pairs of residues,
runs over the aligned pairs of residues, ![]() is the distance between the
is the distance between the ![]() such pair, and
such pair, and ![]() is a normalization term that approximates the average distance between two randomly chosen points in a globular protein of length
is a normalization term that approximates the average distance between two randomly chosen points in a globular protein of length ![]() [Zhang and Skolnick, 2004]. A perfect alignment has a TM-score of 1.0, and two proteins with a TM-score
[Zhang and Skolnick, 2004]. A perfect alignment has a TM-score of 1.0, and two proteins with a TM-score ![]() 0.5 are often said to show structural homology [Xu and Zhang, 2010].
0.5 are often said to show structural homology [Xu and Zhang, 2010].
The Align Protein Structure Algorithm attempts to find the structure alignment with the highest TM-score. This problem reduces to finding a sequence alignment that pairs residues in a way that results in a high TM-score. Several sequence alignments are tried including an alignment with the BLOSUM62 matrix, an alignment of secondary structure elements, and iterative refinements of these alignments.
The Align Protein Structure Algorithm is also capable of aligning entire protein complexes. To do this, it must determine the correct pairing of each chain in one complex with a chain in the other. This set of chain pairings is determined by the following procedure:
- Make structure alignments between every chain in one complex and every chain in the other. Discard pairs of chains that have a TM-score of
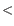 0.4
0.4
- Find all pairs of structure alignments that are consistent with each other i.e. are achieved by approximately the same rotation
- Use a heuristic to combine consistent pairs of structure alignments into a single alignment
The heuristic used in the last step is similar to that of MM-align [Mukherjee and Zhang, 2009], whereas the first two steps lead to both a considerable speed up and increased accuracy. The alignment of two 30S ribosome subunits, each with 20 protein chains, can be achieved in less than a minute (PDB codes 2QBD and 1FJG).