K-mer Based Tree Construction
The "K-mer Based Tree Construction" uses single sequences or sequence lists as input and is the simplest way of creating a distance-based phylogenetic tree. To run the "K-mer Based Tree Construction" tool:
Toolbox | Classical Sequence Analysis (![]() ) | Alignments and Trees (
) | Alignments and Trees (![]() )| K-mer Based Tree Construction (
)| K-mer Based Tree Construction (![]() )
)
Select sequences or a sequence list (figure 21.1):
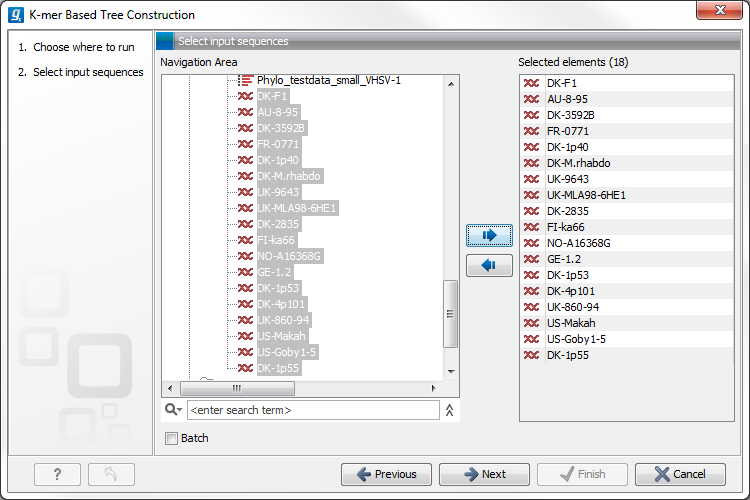
Figure 21.1: Creating a tree with K-mer based tree construction. Select sequences.
Next, select the construction method, specify the k-mer length and select a distance measure for tree construction (figure 21.2):
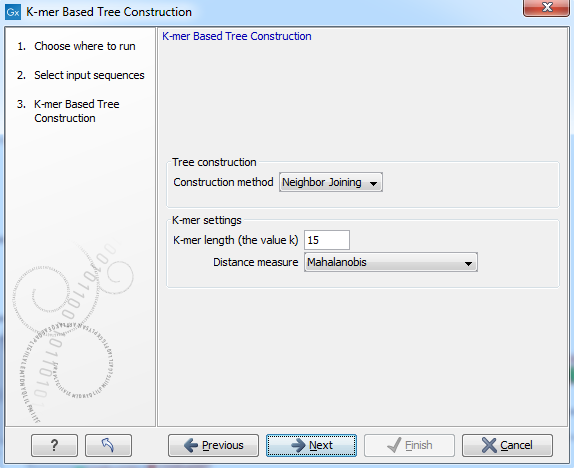
Figure 21.2: Creating a tree with K-mer based tree construction. Select construction method, specify the k-mer length and select a distance measure.
- Tree construction
- Tree construction method The user is asked to specify which distance-based method to use for tree construction.
There are two options (see Distance-based methods):
- The UPGMA method. Assumes constant rate of evolution.
- The Neighbor Joining method. Well suited for trees with varying rates of evolution.
- Tree construction method The user is asked to specify which distance-based method to use for tree construction.
There are two options (see Distance-based methods):
- K-mer settings
- K-mer length (the value k) Allows specification of the k-mer length, which can be a number between 3 and 50.
- Distance measure The distance measure is used to compute the distances between two counts of k-mers. Three options exist: Euclidian squared, Mahalanobis, and Fractional common K-mer count. See 21.2.5 for further details.