SOLiD from Life Technologies
Choosing the SOLiD import will open the dialog shown in figure 6.8.
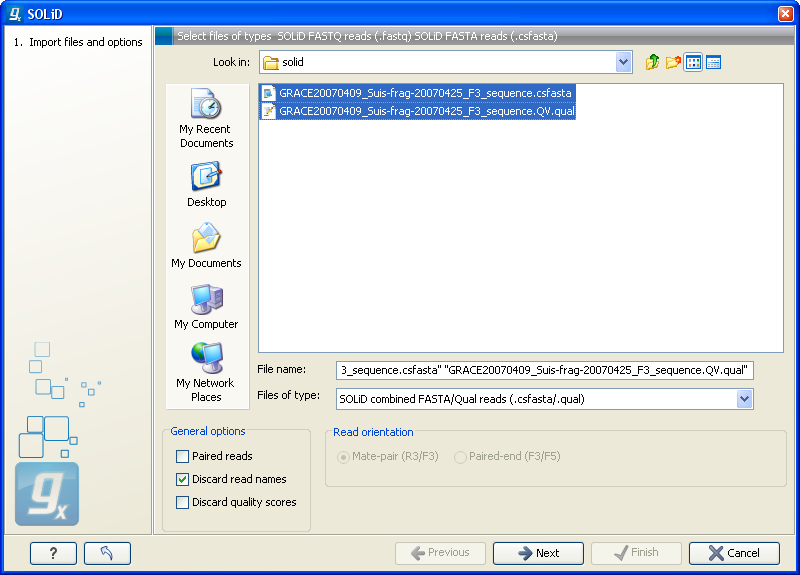
Figure 6.8: Importing data from SOLiD from Applied Biosystems.
There are two formats accepted: the XSQ format which is the native format of newer SOLiD systems, and the csfasta format which is the color space version of fasta format.
The XSQ format
An XSQ file can contain results from multiple libraries produced from the same sequencing run. These are identified by a barcode on each read, and when the XSQ file is produced, each read is placed into its appropriate library based on its barcode. The XSQ importer creates separate sequence lists for each library.
Sometimes when an XSQ file is produced a barcode can not be identified accurately enough to place the read into a specific library, or the read is for some other reason not assigned to a library. In this case, the read is placed into an "Unclassified" or "Unassigned" library.
In the case of paired reads, it sometimes happens that one read of a pair could not be read. When the XSQ file is imported in the CLC Genomics Workbench, the other read of such a pair is placed into a sequence list with " (single)" appended to the name, whereas all intact pairs are placed (alternating) into a sequence list with " (paired)" appended to the name. Thus, two sequence lists are produced for the library.
Hence, when importing data in XSQ format the number of imported files can vary. In the example shown here, where the XSQ file contain a library with the name "Main" (containing paired reads) and an "Unclassified" library (containing reads where e.g. the barcode could not be read), the imported data are segregated into the following sequence lists:
- Main (single)
- Main (paired)
- Unclassified
The csfasta format
If you want to import quality scores with csfasta files, qual files should also be provided. The reads in a csfasta file look like this:
>2_14_26_F3 T011213122200221123032111221021210131332222101 >2_14_192_F3 T110021221100310030120022032222111321022112223 >2_14_233_F3 T011001332311121212312022310203312201132111223 >2_14_294_F3 T213012132300000021323212232.03300033102330332All reads start with a T which specifies the right phasing of the color sequence.
If a reads has a . as you can see in the last read in the example above, it means that the color calling was ambiguous (this would have been an N if we were in base space). In this case, the Workbench simply cuts off the rest of the read, since there is no way to know the right phase of the rest of the colors in the read. If the read starts with a dot, it is not imported. If all reads start with a dot, a warning dialog will be displayed. The handling of dots is identical for XSQ and csfasta files.
In the quality file, the equivalent value is -1, and this will also cause the read to be clipped.
When the example above is imported into the Workbench, it looks as shown in figure 6.9.
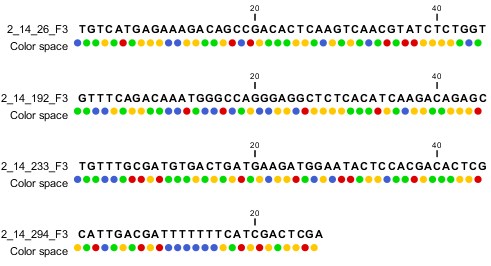
Figure 6.9: Importing data from SOLiD from Applied Biosystems. Note that the fourth read is cut off so that the color following the dot are not included
For more information about color space, please see Color space.
In addition to the csfasta and XSQ formats used by SOLiD, you can also input data in fastq format. This is particularly useful for data downloaded from the Sequence Read Archive at NCBI (http://www.ncbi.nlm.nih.gov/Traces/sra/). An example of a SOLiD fastq file is shown here with both quality scores and the color space encoding:
@SRR016056.1.1 AMELIA_20071210_2_YorubanCGB_Frag_16bit_2_51_130.1 length=50 T31000313121310211022312223311212113022121201332213 +SRR016056.1.1 AMELIA_20071210_2_YorubanCGB_Frag_16bit_2_51_130.1 length=50 !*%;2'%%050%'0'3%%5*.%%%),%%%%&%%%%%%'%%%%%'%%3+%%% @SRR016056.2.1 AMELIA_20071210_2_YorubanCGB_Frag_16bit_2_51_223.1 length=50 T20002201120021211012010332211122133212331221302222 +SRR016056.2.1 AMELIA_20071210_2_YorubanCGB_Frag_16bit_2_51_223.1 length=50 !%%)%'))'&'%(((&%/&)%+(%%%&%%%%%%%%%%%%%%%+%%%%%%+'
For all formats, compressed data in gzip format is also supported (.gz).
The General options to the left are:
- Paired reads. When you import paired data, two different protocols are supported:
- Mate-pair. For mate-pair data, the reads should be in two files with
_F3and_R3in front of the file extension. The orientation of the reads is expected to be forward-forward. - Paired-end. For paired-end data, the reads should be in two files with
_F3and_F5-P2or_F5-BC. The orientation is expected to be forward-reverse.
Read more about handling paired data. Please note that for XSQ files, the pairing protocol is defined in the file itself, which means that the choices of protocol will be ignored.
An example of a complete list of the four files needed for a SOLiD mate-paired data set including quality scores:
dataset_F3.csfasta dataset_F3.qual dataset_R3.csfasta dataset_R3.qual
or
dataset_F3.csfasta dataset_F3_.QV.qual dataset_R3.csfasta dataset_R3_.QV.qual
- Mate-pair. For mate-pair data, the reads should be in two files with
- Discard read names. For high-throughput sequencing data, the naming of the individual reads is often irrelevant given the huge amount of reads. This option allows you to discard this option to save disk space.
- Discard quality scores. Quality scores are visualized in the mapping view and they are used for SNP detection. If this is not relevant for your work, you can choose to Discard quality scores. One of the benefits from discarding quality scores is that you will gain a lot in terms of reduced disk space usage and memory consumption. If you choose to discard quality scores, you do not need to select a .qual file when importing csfasta files.
Click Next to adjust how to handle the results. We recommend choosing Save in order to save the results directly to a folder, since you probably want to save anyway before proceeding with your analysis. There is an option to put the import data into a separate folder. This can be handy for better organizing subsequent analysis results and for batch processing.