Running the QIAseq UPXome workflows
When running the workflow, a barcode reference table must be supplied. It can be either:
- the list of barcodes generated from the Detect Wells tool described previously;
- the reference data element from the QIAseq UPXome and FastSelect RNA hg38 QIAGEN sets located in the References manager.
All QIAseq UPXome workflows are designed to analyze all samples within an experiment without the need of batch functionality.
To run a workflow, first select the input reads, see figure 14.13.
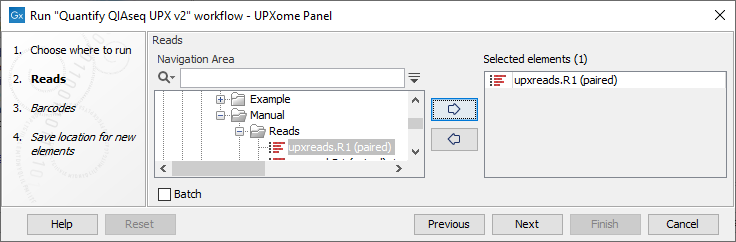
Figure 14.13: QIAseq UPXome workflows require multiplexed reads and analyze all samples within an experiment without the need of batch functionality.
Next, select the wells used in the experiment. First select the barcode table that will populate the dialog, see figure 14.14.
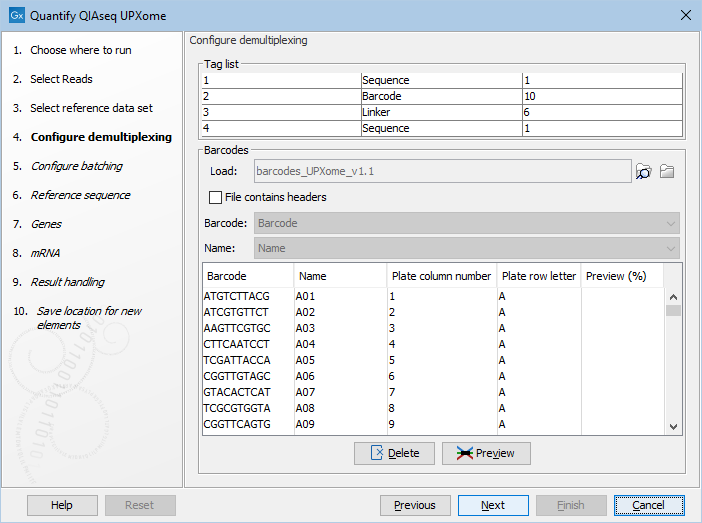
Figure 14.14: Load or import barcodes to have them visualized in the dialog.
Use Preview to identify relevant wells, click the "Preview (%)" column to sort by the number of reads per well, and Delete empty wells that are not of interest, see figure 14.15. In this way it is possible to tailor the number of elements generated to match the experimental design.
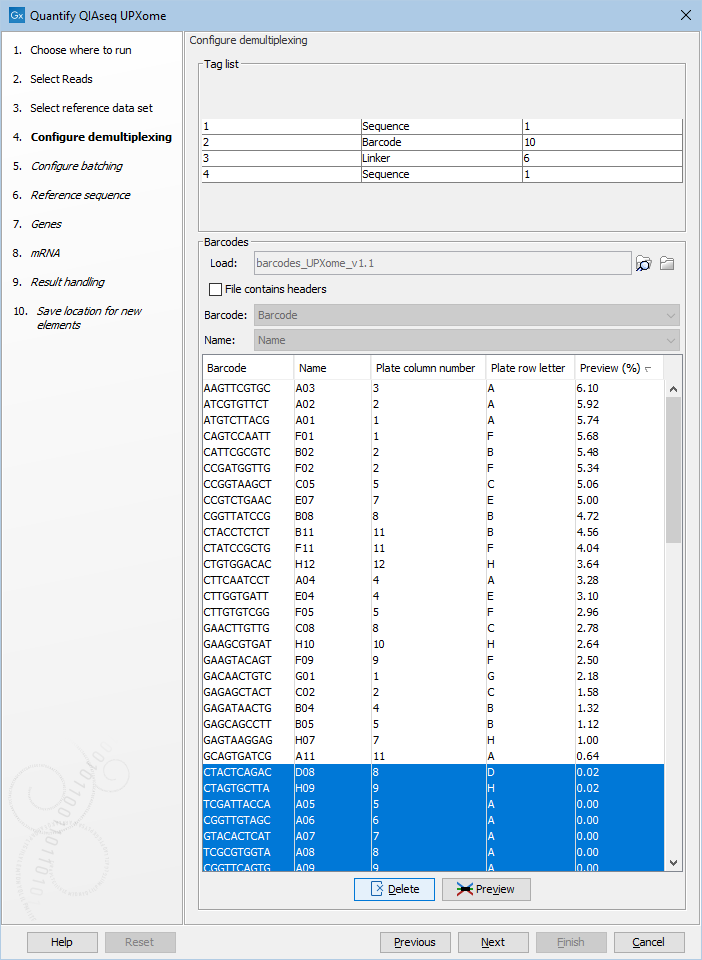
Figure 14.15: Use preview, then sort the barcodes and delete the unused wells.
Finally, specify the save location for the workflow output.
Launching using the QIAseq Panel Analysis Assistant
The workflows are also available in the QIAseq Panel Analysis Assistant under UPXome RNA.
The barcodes are taken from the QIAseq UPXome reference data set and there is no step to supply the barcodes.
When running a workflow from the QIAseq Panel Analysis Assistant the well selection step looks like figure 14.16. The Detect Wells tool (see Detect Wells) can be used to get a list of wells detected using the first 10,000 reads of the input.
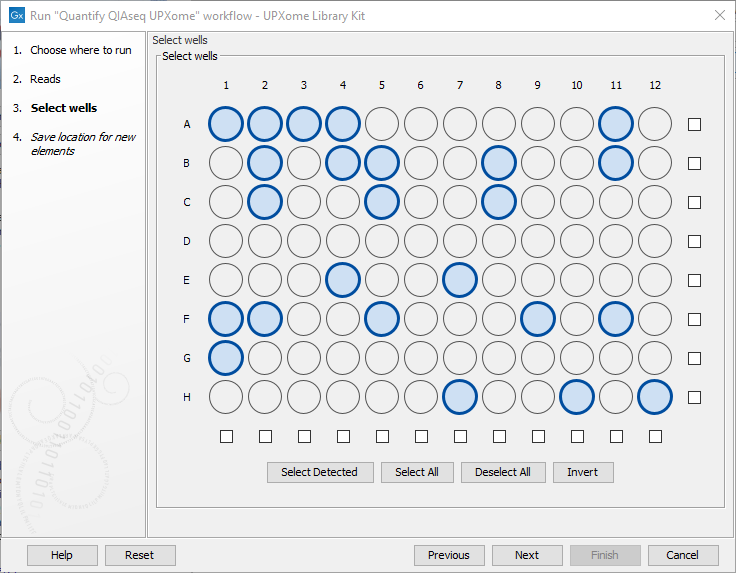
Figure 14.16: The wells used in the experiment should be selected in this step.