Trim Primers of Mapped Reads
The tool Trim Primers of Mapped Reads removes the primer parts of mapped reads, as they reflect the primer that was added and not the actual sample. Note that the tool will also remove any insertion located immediately after the primer. The tool also removes artifacts due to mispriming events, happening when a primer binds an off-target location and thus amplifies an off-target sequence.
Trim Primers of Mapped Reads is available under the Tools menu at:
Tools | Biomedical Genomics Analysis (![]() ) | Biomedical Utility Tools (
) | Biomedical Utility Tools (![]() ) | Trim Primers of Mapped Reads (
) | Trim Primers of Mapped Reads (![]() )
)
In the first dialog (figure 6.10), select a read mapping.
Figure 6.10: Select a read mapping.
In the second dialog (figure 6.11), select the primer annotation track that was provided with the QIAseq Panel. This track contains the original primers and their intended primer locations.
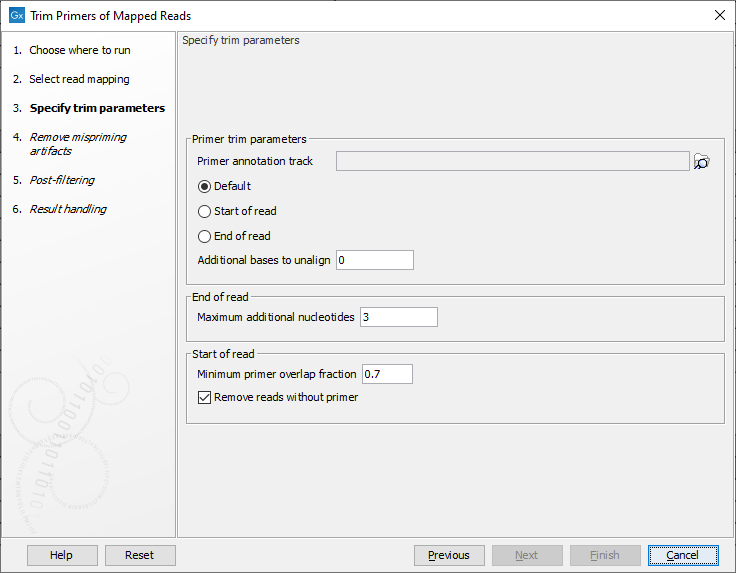
Figure 6.11: Select the primer annotation track specific to the panel, and add the parameters needed to deal with the type of reads you are working with.
In addition, set the following parameters:
- Primer location
- Default: primers are at the end of single-end reads, or at the start of read 1 for paired-end reads.
- Start of read: primers are at the start of single-end reads, or at the start of read 1 for paired-end reads.
- End of read: primers are at the end of single-end reads. This option is not supported for paired-end reads.
- Start of read 2: primers are at the start of read 2 of paired-end reads. This option is not supported for single-end reads.
- Decide on how many Additional bases to unalign immediately after the primer. This trimming is not done on reads for which dimer artifacts are identified.
- Maximum additional nucleotides: When trimming primers from the end of single-end reads, unalign reads that end up to this number of extra bases after the primer.
- Minimum primer overlap fraction: If an aligned read starts within the span of a primer, and if it overlaps at least this percentage of the primer, then it is said to "hit" the primer. For reads "hitting" a primer, the part of the read that overlaps the primer will be unaligned.
- Remove reads without primer When enabled, reads not "hitting" a primer and not coming from broken pairs will be removed from the output mapping. Broken pair reads are retained to help visualize genomic rearrangements. Note that it is possible to later remove broken pairs from the output mapping by running the tool Extract Reads with the option "Include reads from broken pairs" deselected.
If one read in a UMI group runs past the primer it overlaps, it means that all reads in that group were not created from that primer. If this happens, then the tool will not unalign any reads in this UMI group.
In the Remove mispriming artifacts dialog (figure 6.12), you can specify a Primer mispriming events track containing the predicted off-target priming locations of the original primers. Mispriming tracks can be generated by the tool Identify Mispriming Events (see section 6.3) and are available for each QIAseq panel from the Reference Data Manager.
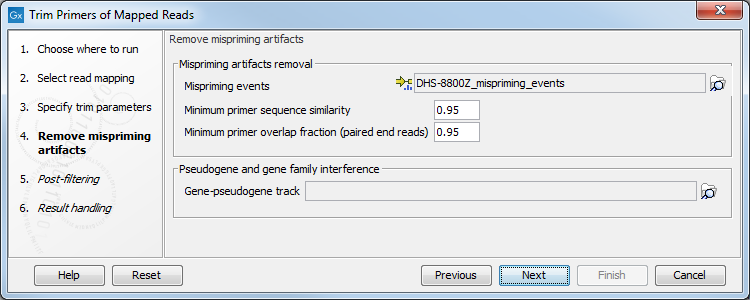
Figure 6.12: Select a mispriming track specific to the panel, and configure the associated parameters if needed.
In addition, set the following parameters:
- Mispriming artifacts removal. The tool will unalign primer part of misprimed reads only if the misprimer overlapping part of the read has at least the Minimal primer sequence similarity fraction to the original primer sequence. For paired end reads, the tool will unalign primer of misprimed paired end reads only if the primer part has at least the Minimal primer overlap fraction with the off-target primer.
- Pseudogene and gene family interference. It is possible to specify a gene-pseudogene track that contains gene and pseudogene links information for removing reads that map well to pseudogene locations.
In the Post-filtering dialog (figure 6.13), when the "Remove short reads" option is enabled, reads with an alignment length shorter than the value specified after primer trimming will be removed from the mapping.
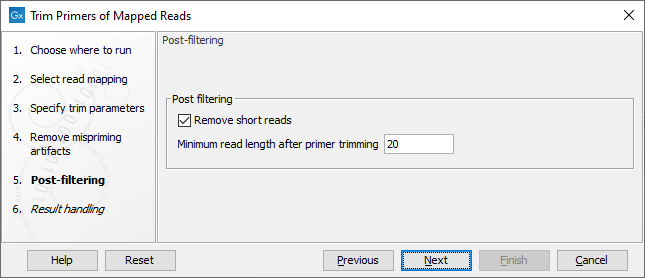
Figure 6.13: Post-filtering parameters.
The tool will output a trimmed read mapping. In addition, as seen in figure 6.14, you can choose to output off-target primers and/or regions tracks with counts.
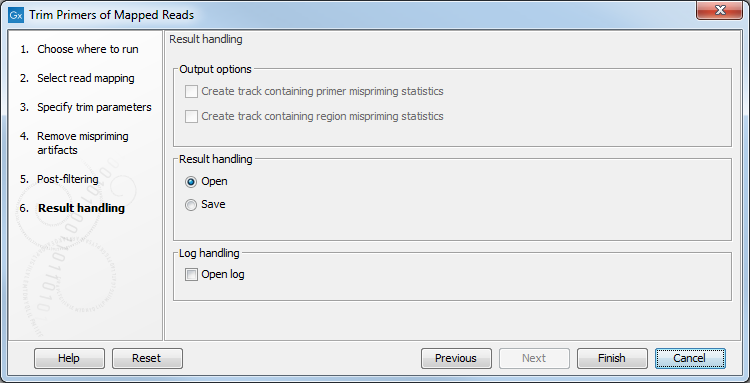
If a mispriming track was specified in the earlier step, it is possible to output two additional tracks containing respectively region and primer mispriming statistics:
- The primer mispriming statistics track (figure 6.15) includes mispriming events and the counts of read unalignment those events caused in a process of mispriming artifact removal.
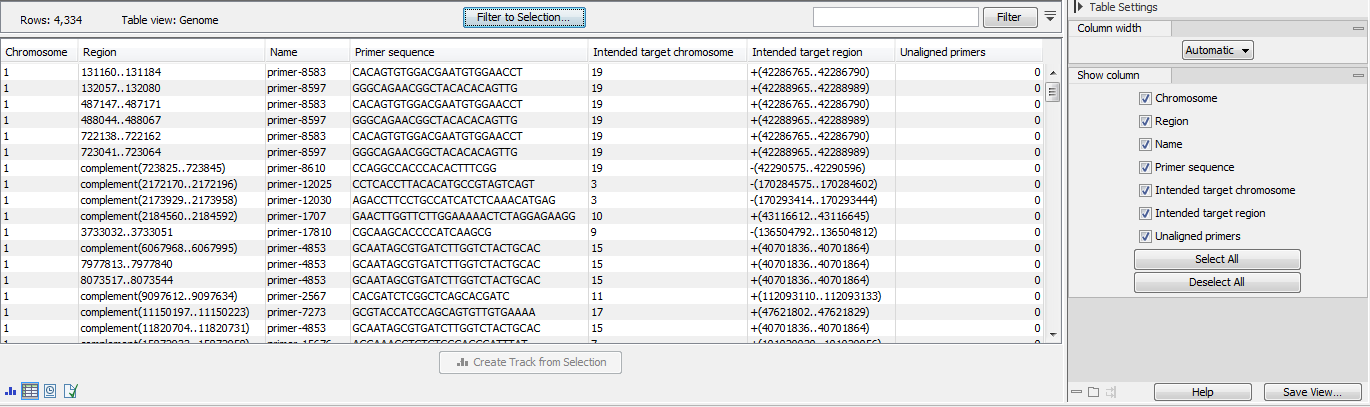
Figure 6.15: A primer mispriming track.- Chromosome, Region and Name referring to the identified mispriming locations.
- Intended target chromosome and
- Intended region referring to the true location of the primer.
- Unaligned primers Total number of reads supporting the mispriming event. These reads are amplified from a mispriming event, and the primer part of the read has been unaligned because it matched the primer sequence and region. The primer part of the read might contain mismatches if the primer doesn't match the reference exactly. It is only unaligned if both the sequence and region overlap is sufficient, as determined by the two input parameters, "Minimum primer sequence similarity" and "Minimum primer overlap fraction (paired end reads)". For single end reads, only the first parameter is applicable.
- The regions mispriming statistics track (figure 6.16) includes regions that are considered as alternative mapping locations when attempting to remove mismapped reads. Primary target locations in this track are annotated with:
- Name of the primer
- Primary target True or false depending if the region describes the primer or a possible mispriming event.
- Reads checked Total number of reads checked for better matches to a mispriming event region or to a position in the vicinity of the original mapping location.
- Reads matching mispriming location Number of reads mapping with a better score to a mispriming region than to the original location after the primer sequence had been unaligned. These reads are removed from the read mapping.
- Reads matching target vicinity Number of reads with a better match to a position in the vicinity of the original mapping location after the primer sequence had been unaligned. These reads are removed from the read mapping.
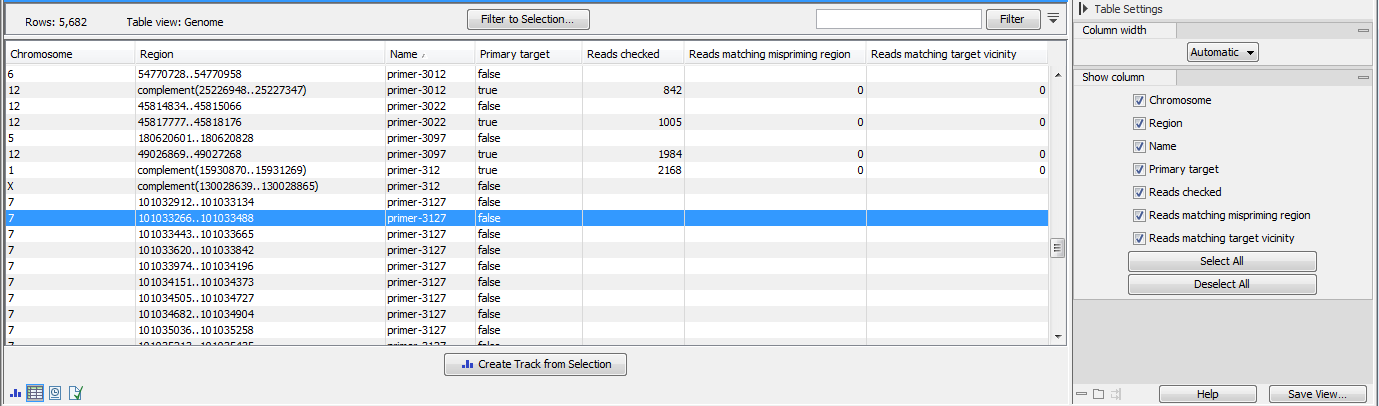
Figure 6.16: A region mispriming track.