Output from the Filter Somatic Variants (TAS) workflow
Two types of output are generated:
- Amino Acids Changes Track that shows the consequences of the variants at the amino acid level in the context of the original amino acid sequence. A variant introducing a stop mutation is illustrated with a red amino acid.
- Somatic Candidate Variants Track that holds the variant data. This track is also included in the Track List. If you hold down the Ctrl key (Cmd on Mac) while clicking on the table icon in the lower left side of the View Area, you can open the table view in split view. The table and the variant track are linked together, and when you click on a row in the table, the track view will automatically bring this position into focus.
- Track List Filter Somatic Variants A collection of tracks presented together. Shows the somatic candidate variants together with the human reference sequence, genes, transcripts, coding regions, and variants detected in ClinVar, 1000 Genomes, and the PhastCons conservation scores (see figure 20.18).
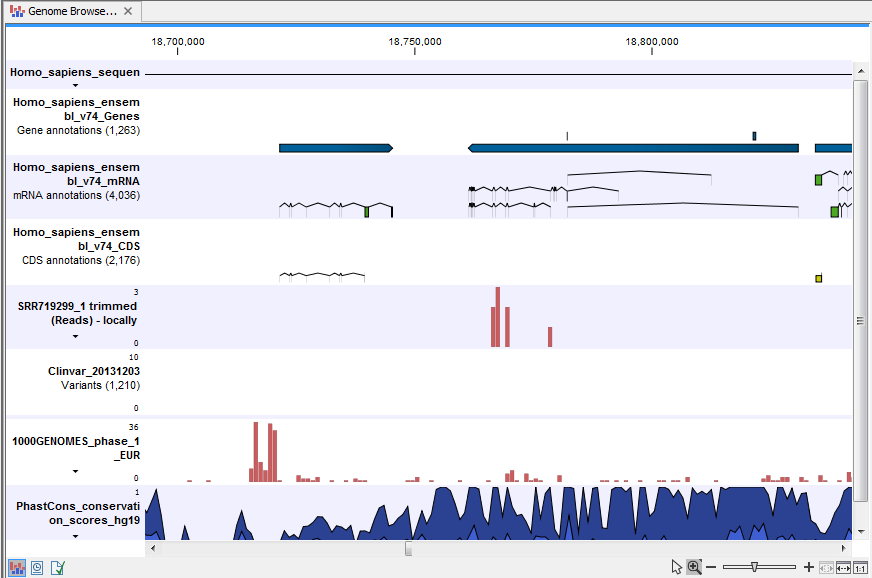
Figure 20.18: The Track List showing the annotated somatic variants together with a range of other tracks.
To see the level of nucleotide conservation (from a multiple alignment with many vertebrates) in the region around each variant, a track with conservation scores is added as well. Mapped sequencing reads as well as other tracks can be easily added to this Track List. Open the variant track as a table showing all variants and the added information/annotations (see figure 20.19).
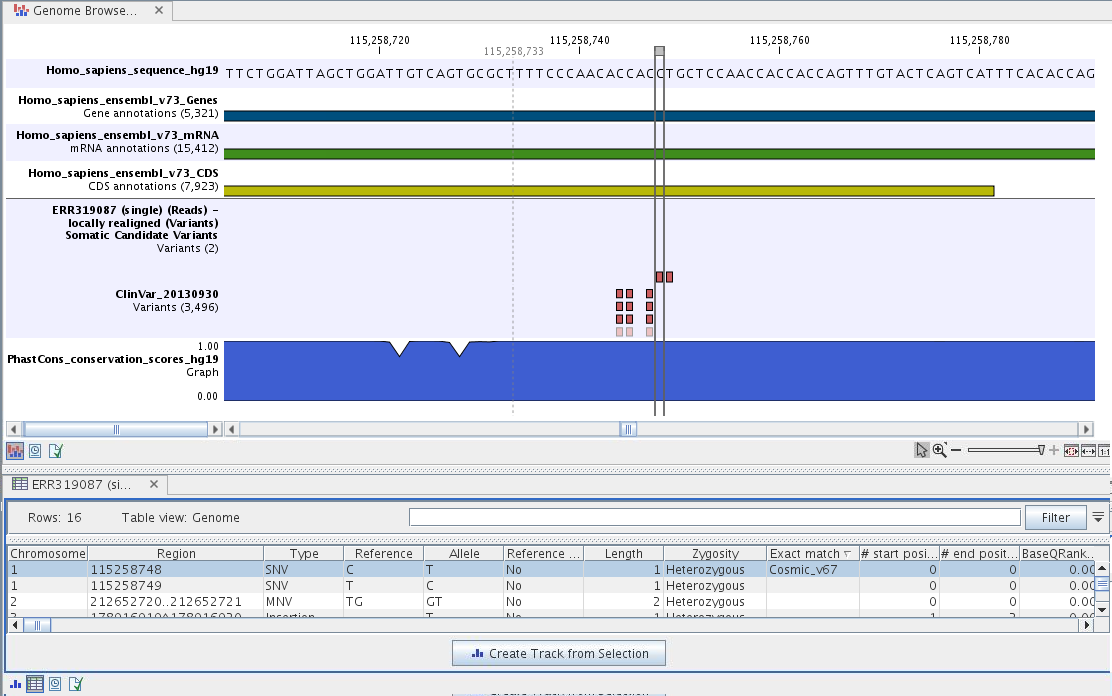
Figure 20.19: The Track List showing the annotated somatic variants together with a range of other tracks.
Adding information from other sources may help you identify interesting candidate variants for further research. E.g. common genetic variants (present in the HapMap database) or variants known to play a role in drug response or other relevant phenotypes (present in the ClinVar database) can easily be identified. Further, variants not found in the ClinVar databases, can be prioritized based on amino acid changes in case the variant causes changes on the amino acid level.
A high conservation level between different vertebrates or mammals, in the region containing the variant, can also be used to give a hint about whether a given variant is found in a region with an important functional role. If you would like to use the conservation scores to identify interesting variants, we recommend that variants with a conservation score of more than 0.9 (PhastCons score) is prioritized over variants with lower conservation scores.
It is possible to filter variants based on their annotations. This type of filtering can be facilitated using the table filter found at the top part of the table. If you are performing multiple experiments where you would like to use the exact same filter criteria, you can include in a workflow the Filter on Custom Criteria tool configured with the desired set of criteria.