Tables for Clonotype Sample Comparison
Clonotype Sample Comparisons have two table views:
- Clonotypes (
 ). If the same clonotype is present in multiple samples, there will be just one row for the clonotype. The Samples column contains the number of samples in which the clonotype was identified.
). If the same clonotype is present in multiple samples, there will be just one row for the clonotype. The Samples column contains the number of samples in which the clonotype was identified.
- Clonotypes on Sample Level (
 ). If the same clonotype is present in multiple samples, there will be one row for each sample. The Sample column contains the sample where the clonotype was identified.
). If the same clonotype is present in multiple samples, there will be one row for each sample. The Sample column contains the sample where the clonotype was identified.
Both tables have the following columns:
- Chain:
Which chain type the clonotype belongs to: TRA (
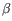 ), TRB (
), TRB (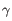 ), TRG (
), TRG (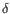 ) and TRD (
) and TRD (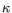 ) for TCR, or IGH (heavy), IGK (light
) for TCR, or IGH (heavy), IGK (light 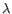 ), and IGL (light
), and IGL (light 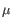 ) for BCR. Note that other light BCR chain types are currently not supported.
) for BCR. Note that other light BCR chain types are currently not supported.
- V, D, J and C: The identified V, D, J and C reference segment(s). If a single unambiguous V / D / J / C segment could not be identified, the segments will be listed separated by a comma.
- CDR3 nucleotide sequence: The nucleotide sequence for CDR3 including the V and J segment-encoded conserved amino acids.
- CDR3 amino acid sequence: The translated amino acid sequence for the CDR3 nucleotide sequence provided that it is in-frame.
- CDR3 length: The length of the CDR3 nucleotide sequence.
- Count: The number of fragments for which the specific clonotype was detected. A fragment represents one single read or one pair of reads.
- Frequency (%): The count given as a percentage relative to the sum of all counts. Note that filtering can affect frequencies, see Filter Immune Repertoire.
- Productive:
One of three categories are used to characterize the CDR3 nucleotide sequences:
- Productive: sequences that are in frame and do not contain a premature stop codon;
- Out-of-frame: sequences that have a length that is not a multiple of three;
- Premature stop codon: sequences that are in-frame but contain a premature stop codon.
The Clonotypes view (![]() ) has one Count and one Frequency (%) column for each sample.
) has one Count and one Frequency (%) column for each sample.
Clonotype Sample Comparison from selection
At the bottom of each table, a button labeled Create Comparison from Selection is available. Select the relevant rows in the table and click the button to create a new Clonotype Sample Comparison (![]() ) that only includes the selected clonotypes. When the button is clicked, a dialog with the following options is shown:
) that only includes the selected clonotypes. When the button is clicked, a dialog with the following options is shown:
- Recalculate frequencies. If ticked, frequencies in the output clonotypes are recalculated such that they add up to 100% across all chains. Otherwise, the original frequencies found in the input are used.
It can be useful to recalculate frequencies when removing noise (for example, removing clonotypes with a count of 1), but if a subset of clonotypes is created for the purpose of comparing clonotypes between samples, it might be more relevant to preserve the original frequencies.
- Set frequencies per chain. If ticked, the frequencies are recalculated to add up to 100% for each individual chain. This option is enabled only when Recalculate frequencies is ticked.
Note, that the frequencies are always recalculated separately for each sample in the selection.