Perform TSO500 RNA Analysis
The Perform TSO500 RNA Analysis (Illumina) workflow includes all necessary steps for processing paired-end reads from TSO500 RNA samples, such as sample QC, adapter trimming, mapping resulting in gene expression counts and fusion gene identification.
This template workflow is available under the Workflows menu at:
Workflows | Template Workflows | Biomedical Workflows (![]() ) | TSO Panel Analysis (
) | TSO Panel Analysis (![]() ) | Perform TSO500 RNA Analysis (Illumina) (
) | Perform TSO500 RNA Analysis (Illumina) (![]() )
)
If you are connected to a CLC Server via the CLC Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the next step, select the RNA sequencing reads to analyze. The input can be sequence lists containing paired-end reads selected from the Navigation Area, or samples can be imported using the "Select files for on-the-fly import" option, where files containing paired-end read data can be selected from disk. If choosing the "Select files for on-the-fly import" option, the "Paired reads" option needs to be enabled.
If you would like to analyze more than one sample in one workflow run, check the "Batch" box in the lower left corner of the dialog. When analyzing multiple imported samples, metadata needs to be provided. Further information can be found at https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Running_workflows_in_batch_mode.html.
After selecting reference data as described below you can configure the batch unit and see the batch overview. When using metadata, selecting an Excel file that describes the data will often be the most convenient method. Providing metadata directly from an Excel file is the only option available when input data is imported as part of the workflow run.
The following dialog helps you set up the relevant Reference Data Set. If you have not downloaded the Reference Data Set yet, the dialog will suggest the relevant data set and offer the opportunity to download it using the Download to Workbench button. The dialog for selection of reference data is shown in figure 15.1.
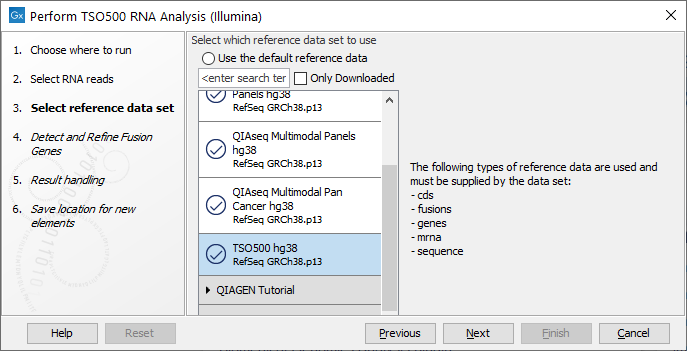
Figure 15.3: The relevant Reference Data Set is highlighted. The text to the right lists the types of references needed by the workflow.
Note that if you wish to Cancel or Resume the Download, you can close the template workflow and open the Reference Data Manager where the Cancel, Pause and Resume buttons are available.
If the Reference Data Set was previously downloaded, the option "Use the default reference data" is available and will ensure the relevant data set is used. You can always check the "Select a reference set to use" option to be able to specify another Reference Data Set than the one suggested.
In the next step, you can adjust the parameters for specifying the fusion gene detection (figure 15.4).
The Configurable Parameters for Detect and Refine Fusion Genes are:
- Promiscuity threshold: The number of genes a fusion gene is allowed to fuse with. We recommend to keep this value under 10. The higher this value is, the more potential false positives can be produced. The likelihood for true fusion genes to be able to fuse with multiple genes is not that high. Genes with a high promiscuity rate are often ribosomal genes which are more likely to have a long polyA tail that can fuse in different places and thereby indicate potential gene fusions. This is especially true when the "Detect fusion with novel exon boundaries" is enabled, as this option allows fusion to intronic regions where polyA stretches are more prominent. However, some genes do tend to fuse in intronic regions creating novel exons such as PLM-RARA where this phenomenon is often seen.
- Detect exon skippings: Allows for detection of novel transcripts that origin from exon skipping events.
- Detect fusions with novel exon boundaries: When enabled fusions with breakpoints that are within a specified distance to the known exon boundary, but not at canonical exon boundaries, are also reported. The default setting for "Maximum distance to known exon boundary" is 8.
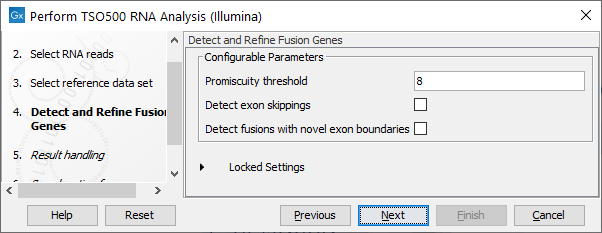
Figure 15.4: Adjustable parameters for the Detect and Refine Fusion Genes wizard step. Options include detection of exon skipping and fusions with novel exon boundaries.
Finally, choose where to save the results.
Subsections