Convert Annotation Track Coordinates
Convert Annotation Track Coordinates converts annotation coordinates, either from hg19 coordinates to hg38 coordinates, or vice versa. This remapping of coordinates, also referred to as 'liftover', makes use of the UCSC Lift Genome Annotations service (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Network access is therefore necessary to use this tool.
The assemblies used for remapping are hg19 (Feb. 2009 (GRCh37/hg19)) and hg38 (Dec. 2013 (GRCh38/hg38)).
Limitations:
- It is possible to remap annotation coordinates to and from the reference sequence hg38_no_alt_analysis_set, but annotations cannot be remapped to or from the alternative contigs that are included in hg38_no_alt_analysis_set.
- The tool can only convert files that are under 500Mb.
The UCSC Lift Genome Annotations service provides a list of options that can be adjusted. Of those, only Minimum ratio of bases that must remap is available in the Convert Annotation Track Coordinates tool. The remaining options are set to default values:
- BED 4 to BED 6 Options
- Allow multiple output regions - Checked
- Minimum hit size in query - 0
- Minimum chain size in target - 0
- BED 12 Options
- Minimum ratio of alignment blocks or exons that must map - 1
- If thickStart/thickEnd is not mapped, use the closest mapped base - unchecked
To run Convert Annotation Coordinates Track, go to:
Tools | Biomedical Genomics Analysis (![]() ) | Biomedical Utility Tools (
) | Biomedical Utility Tools (![]() ) | Convert Annotation Track Coordinates (
) | Convert Annotation Track Coordinates (![]() )
)
The tool takes annotation tracks, typically primer tracks or target region tracks, as input (see figure 6.17).
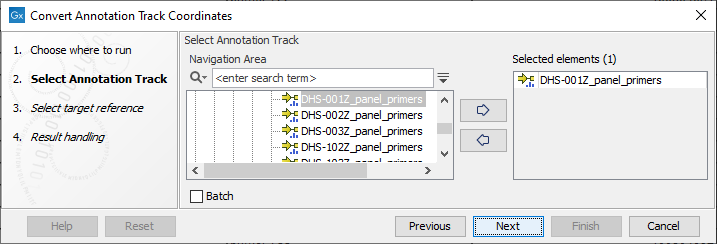
Figure 6.17: Selection of a primer annotation track with hg19 coordinates for conversion to hg38 coordinates.
In the next step, a target reference must be selected (see figure 6.18). If the annotation track used as input was based on an hg19 reference, select an hg38 reference genome. If the annotation track used as input was based on an hg38 reference, select an hg19 reference genome. If not already available, a copy of the relevant target reference can be downloaded using the Reference Data Manager.
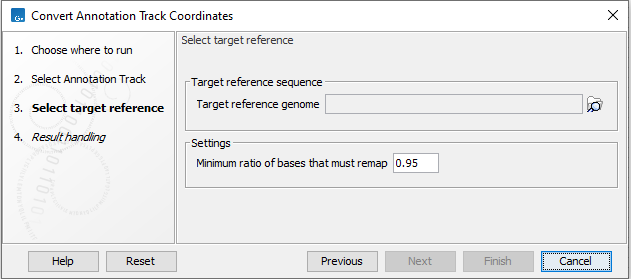
Figure 6.18: This dialog allows selection of the reference sequence you would like to lift over to. Under settings you can choose to adjust the minimum ratio of bases that must remap.
Under settings you can adjust Minimum ratio of bases that must remap. This is the minimum fraction of bases in a region that must be directly aligned in gapless blocks from the first genome to the second.
Convert Annotation Track Coordinates outputs an annotation track containing remapped annotations and a report. The report lists the skipped intervals, that is, the annotations that could not be remapped, together with a comment provided by UCSC (figure 6.19).
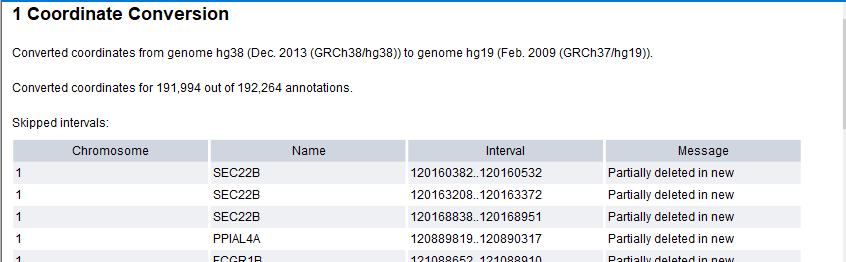
Figure 6.19: The Convert Annotation Track Coordinates report lists skipped intervals.