QIAseq miRNA Quantification
The QIAGEN miRNA Quantification workflow quantifies the expression in a sample of the miRNAs found in miRBase. The workflow includes a Trim Reads step, but note that this step only affects Ion Torrent reads as Illumina reads do not have the 5' adapter.
To run the workflow, go to:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | QIAseq RNA Workflows (
) | QIAseq RNA Workflows (![]() ) |
QIAseq miRNA Quantification (
) |
QIAseq miRNA Quantification (![]() )
)
In the first dialog (figure 13.8), specify the reads to analyze. If the reads come from different samples, remember to check the Batch option. The next dialog will allow you to review the batch units.
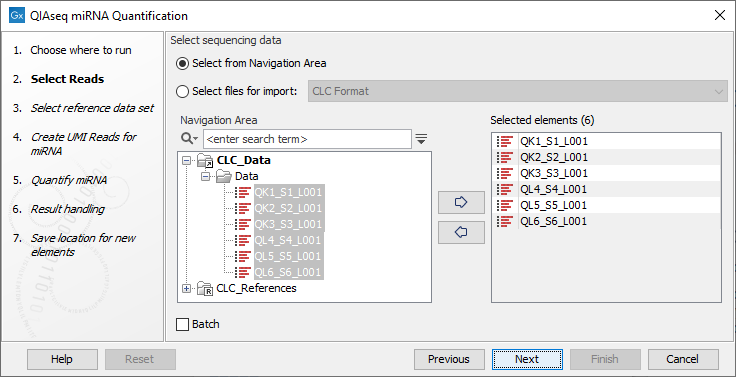
Figure 13.8: Select the reads.
In the dialog shown in figure 13.9, specify the Reference Data Set to be used, for example QIAseq Small RNA when using QIAseq data. This set includes GO annotations, a mapping from miRNA identifiers to GO entries, the miRBase database, spike-ins data and a trim adapter list. Note that alternative data sets can be created as explained in https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Reference_Data_Sets_defining_Custom_Sets.html.
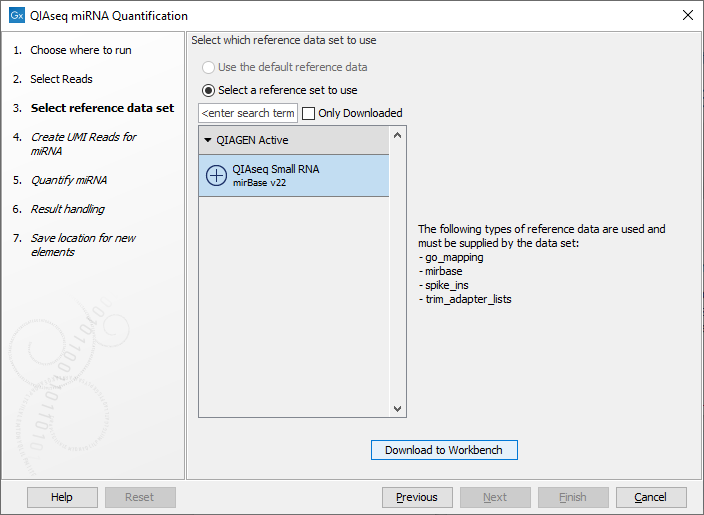
Figure 13.9: Select the relevant Reference Data Set.
In the Create UMI Reads for miRNA dialog (figure 13.10), you can set the parameters needed to run the tool (see Create UMI Reads for miRNA). Note that in this workflow all options have been preconfigured by default to work with Illumina data. How to change the settings when working with Ion Torrent reads is described below.
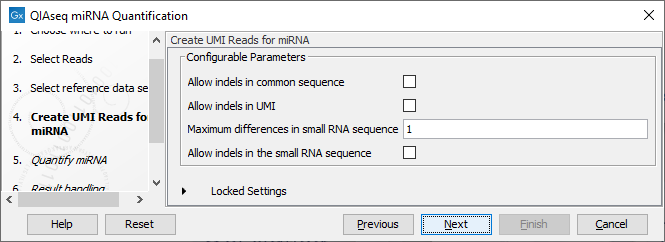
Figure 13.10: Set up the parameters for the Create UMI Reads for miRNA tool.
- Allow indels in common sequence This option is unchecked by default, but should be enabled when working with Ion Torrent data.
- Allow indels in UMI This option is unchecked by default, but should be enabled when working with Ion Torrent data.
- Maximum differences in small RNA sequence Number of allowed differences in the miRNA when merging UMI groups. This is set to 1 difference for Illumina reads, but 2 mismatches should be allowed when working with Ion Torrent data.
- Allow indels in small RNA sequence This option is unchecked by default, but should be enabled when working with Ion Torrent data.
In the Quantify miRNA dialog (figure 13.11), specify the parameters for the tool (see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Quantify_miRNA.html).
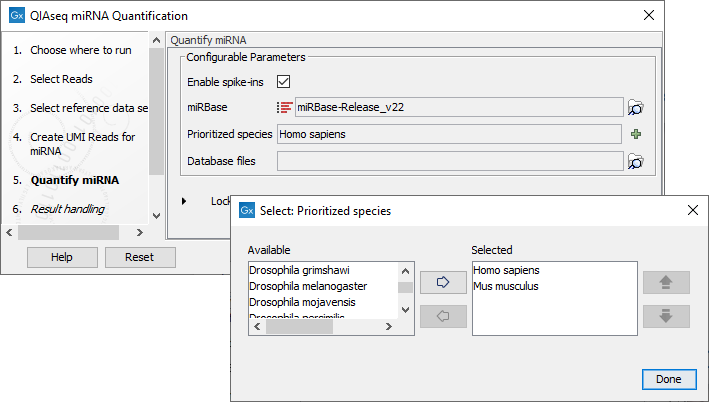
Figure 13.11: Specify the parameters for the Quantify miRNA tool, including the prioritized list of species that should be used to annotate the miRNA.
You can choose whether or not to enable the spike-ins analysis and which miRBase database to use. You can also prioritize a list of species used for annotations, with the actual species sequenced always as the first prioritized. The prioritization is important in cases where a read matches two miRNAs equally well, because only the highest priority match is used when linking the miRNA with GO annotations during gene set testing.
Known limitations of prioritization: when two miRNAs have identical sequences, the prioritization determines which is reported. However, if two miRNAs differ in sequence, prioritization will have no effect. For example, in figure 13.12, species are prioritized in the order `human', `chimpanzee', `mouse'. The sample is a human sample. Nevertheless some reads are assigned to the chimpanzee sequence ptr-miR-143 because they map equally well or better to this miRNA than to human hsa-miR-143-3p.

Figure 13.12: A miRNA will not be prioritized above another when they have different sequences.
In the last step, choose whether to Open or Save your results.
Launching using the QIAseq Panel Analysis Assistant
The workflow is also available in the QIAseq Panel Analysis Assistant under miRNA.
Subsections