Demultiplex QIAseq UPX 3' Reads
The UPX 3' RNA application of the QIAseq Panel Analysis Assistant offers a Demultiplex tool to be run before starting the Analyze QIAseq UPX 3' RNA (Illumina) workflow. The workflow requires the reads to be sequenced with a QIAseq UPX 3' Transcriptome Kit or a QIAseq UPX 3' Targeted RNA Panel.
Click Run to open the wizard. In the first dialog, select the reads to demultiplex, and click Next.
In the following dialog (figure 13.1):
- Select the sequencing platform and the plate size. The initial selections have been automatically inferred. Those selections can be adjusted as needed. (Note: For data from NovaSeq, select the MiSeq/HiSeq as the sequencing platform.)
- Specify whether a mismatch per barcode should be allowed.
- Select all the wells used in the experiment in the diagram in the Select wells area (details below).
Selecting the wells used
A diagram of the plate, based on the selected well size, is shown in the Select wells area. Wells identified automatically as used in the experiment are shaded in blue.
All wells used in the experiment must now be selected. Select wells using functionality associated with the plate diagram, and buttons below it:
- Select a single well by clicking on it in the plate diagram.
- Select individual rows and columns using the checkboxes located to the right and below the diagram.
- Select all wells with the Select All button.
- Deselect all selected wells with the Deselect All button.
- Select only the wells that were automatically detected with the Select Detected button.
- Invert the current selection with the Invert button. Using this button, all selected wells are deselected, and vice versa.
Selected wells are indicated by a dark blue circle around the well. When you click on Next, it is the barcodes for these wells that will be used for demultiplexing.
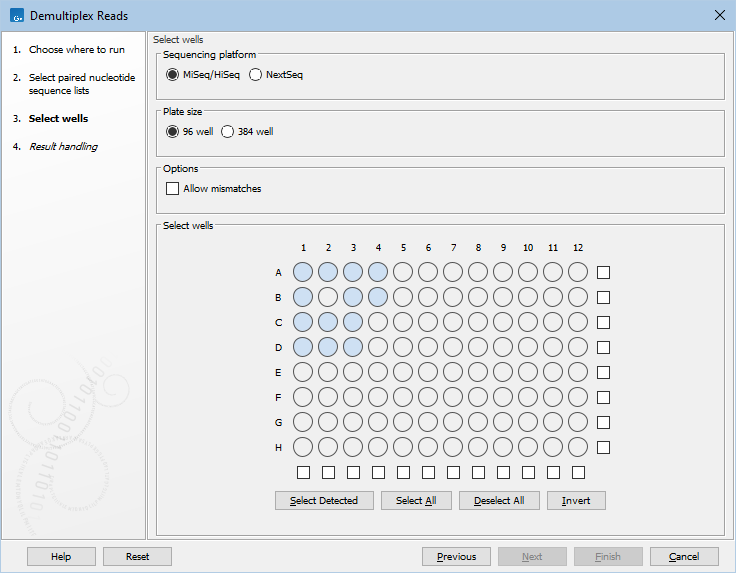
Figure 13.1: Wells identified automatically as being used in the experiment are shaded in light blue. Here, none of the wells have yet been selected for use.
Outputs of demultiplexing
For each sample analyzed, a sequence list is generated for each selected well for which reads have been detected. There are also optional outputs: a sequence list containing reads that did not contain any of the expected barcodes, and a report containing information about the number of reads found for each barcode.
Requirements when running demultiplexing in batch mode
When running demultiplexing in batch mode, all samples being analyzed should have been sequenced on the same platform, and the same wells should have been used for each plate. This is because:
- The wells selected when setting up the analysis will be used for each batch unit.
- The same adapter trimming settings are employed for each batch unit.
Automatic selection of plate size and wells
The automatic selection of plate size and the indication of which wells were used is done by sampling the reads used as input. When using a single file as input, the first 10,000 reads are scanned for barcodes, and this information is used. When multiple files are used as input for a sample, a selection of 10,000 reads is also used, but the selection is taken from across the files.