Validate QIAseq Read Structure (beta)
Validate QIAseq Read Structure (beta) analyzes and validates the read structure for selected QIAseq panels and kits by predicting changes between UMI, common/adapter sequence and biological sequence based on nucleotide contributions for the four DNA nucleotides in the read. It takes a Graphical Report from QC for Sequencing Reads as input.
Validate QIAseq Read Structure (beta) predicts segments in the read structure by analyzing the changes in Standard Deviation (SD) of the nucleotide contributions in a sliding window across the base positions. If the nucleotide contribution for one of the four nucleotides in a position next to the window is significantly different from the nucleotide distribution inside the window, a potential change in read structure is found. The read structure prediction sensitivity parameter determines how many standard deviations the position next to the window should be from the mean of the window, in order to be significantly different.
The tool validates if all predicted segments match the expected read structure. For paired end reads, both R1 and R2 are validated separately.
The validation of read segments is based on three different criteria:
- Start and end positions: The start and end positions of each expected read segment must match the predicted read structure.
- Additional SD changes: If one of the expected read segments contains more than one predicted segment, the difference in nucleotide percentage SD for two adjacent predicted regions must be sufficiently small (by default the difference should be smaller than 10). See figure 5.9 for an example of additional SD changes.
- Nucleotide statistics: Average and SD values of the nucleotide percentage are calculated for each of the expected read segment. At least three of the four nucleotides, A, C, G and T, should have average and SD values that match the type of sequence in the segment. The acceptance criteria for average and SD values in the region depends on the type of sequence:
- UMI: the nucleotides are evenly distributed and the standard deviation should be small (SD < 5). The average value should be between 8 and 50.
- Common sequence: the nucleotides are unevenly distributed and the standard deviation should be high (SD > 8). No requirement for the average value.
- Staggered common sequence: the nucleotides are unevenly distributed and the standard deviation should be high (SD > 6). No requirement for the average value.
- Biological sequence: the nucleotides are evenly distributed however the standard deviation should be small or medium (SD < 20). The average value should be between 12 and 45.
Validate QIAseq Read Structure (beta) is available from the Tools menu at:
Tools | Biomedical Genomics Analysis (![]() ) | QIAseq Tools (
) | QIAseq Tools (![]() ) | Validate QIAseq Read Structure (beta) (
) | Validate QIAseq Read Structure (beta) (![]() )
)
Double-click to run the tool.
Select one or more Graphical Report produced by QC for Sequencing Reads from the navigation area and add them to the list on the right hand side of the dialog using the arrows.
In the next wizard step select the QIAseq protocol used for generating the reads in the 'Expected QIAseq read structure' drop-down menu or select 'Unknown' if the kit is not supported. The tool will be preconfigured with optimal parameters for the different kits, however, sequencing is not an exact science and you might have to adjust the parameters to obtain the expected result. It is possible to adjust a number of parameters, see figure 5.8:
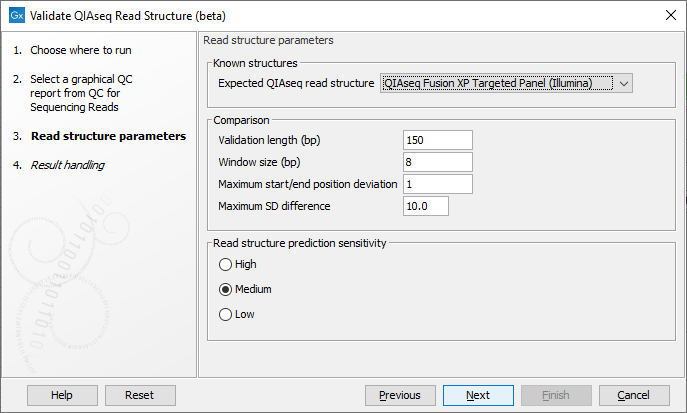
Figure 5.8: Parameter wizard step of the Validate QIAseq Read Structure (beta).
- Comparison parameters:
- Validation length (bp) The number of base positions that are used for validation, counting from the 5' end. Note that in some capture protocols a bias towards a single nucleotide, e.g., polyA-tails, can dominate the end of reads, and in such cases only the stable part of the reads should be included in the analysis.
- Window size (bp) The number of residues considered in the sliding window for calculating standard deviations of the nucleotide percentages. Predicted read segments cannot be shorter than the windows size, so the value should not be smaller than the length of the UMI or common sequence, whichever is shortest. The default value is 8, which allows for more stable SD estimation compared to smaller values, the minimum allowed value is 5.
- Maximum start/end position deviation A segment in the expected read structure fails if the difference between predicted and expected start or end position is larger than this value. Be careful to adjust this value because not all template workflows are build to tolerate a deviation.
- Maximum SD difference A difference larger than this between nucleotide percentage standard deviations for two adjacent predicted regions will be highlighted. Large differences between adjacent regions can indicate a departure from the expected read structure. The standard deviations are calculated as averages over the four DNA nucleotides, hence the SD difference for a single nucleotide could be larger than this value without making the segment fail the comparison.
Note that the different QIAseq protocols have different default values for this parameter, most protocols use 10. The QIAseq Multimodal RNA panel has a default of 30 since the capture protocol results in a polyT-rich region on R2 that will often lead to an unexpected SD change at position
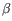 30-32 which should be ignored.
30-32 which should be ignored.
- Read structure prediction sensitivity: The prediction sensitivity affects how sensitive the analysis is with respect to detecting changes in the read structure. The sensitivity might be chosen based on the quality of the reads and/or the sequencing technology. The options are:
- High A high sensitivity results in more read segments being predicted. This setting should be used when sequencing has been performed on the more noisy platforms, e.g., Illuminas MiSeq.
- Medium Default selection that fits most profiles.
- Low A low sensitivity results in fewer read segments being predicted. With this setting, changes in the nucleotide contributions needs to be more significant for splitting the read into segments.
Subsections