Import Immune Reference Segments
Import Immune Reference Segments can import reference sequences for V, D, J and C segments from a fasta file. The sequences are needed when running Immune Repertoire Analysis for either T or B cell receptor repertoires (TCR and BCR, respectively).
The importer can be found here:
Import (![]() ) | Import Immune Reference Segments (
) | Import Immune Reference Segments (![]() ).
).
The following options can be adjusted (figure 7.3):
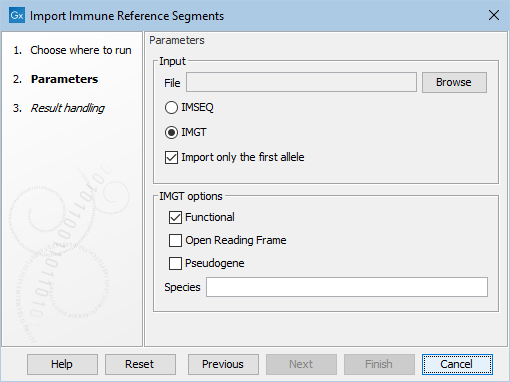
Figure 7.3: The available options when importing immune reference segments.
- File. A fasta file containing the reference sequences.
- File format. Fasta files that use either the format IMSEQ or IMGT can be imported.
The two formats differ in how the sequence header is parsed for identifying the segment type and related information, and how the conserved amino acids in the V and J segments are identified.
- Import only the first allele. Both formats support allele numbering for the gene segments. When this option is checked, only segments without an allele or those with an allele defined as the number "1" (i.e "01" is also valid) are imported. Otherwise, all segments are imported.
- IMGT options can be adjusted when importing using the IMGT format:
- Functionality. Only gene segments with the selected functionalities are imported: Functional, Open reading frame, or Pseudogene.
- Species. Only gene segments from the selected species are imported.
After selecting the fasta file, the desired species can be chosen from those identified within the file.
In the "Result handling" dialog, the reference data for either TCR, BCR, or both, can be saved.
The importer can only import one fasta file at a time, but if more files are imported, the resulting sequence lists can subsequently be combined using Create Sequence List.
Subsections