Sample specific outputs
A separate folder of results is created for each sample and contains sample specific reports, tracks and additional supplemental outputs, (figure 22.7).
Figure 22.7: The per sample outputs generated by the SARS-CoV-2 Low Frequency and Shared Variants workflow
Reports
The following reports are produced:
- trim_report: Reports read lengths before and after trimming, as well as the number of reads discarded because they did not pass the minimum length threshold of 50bp after being trimmed.
- ligation_artifacts_report: Summarizes any ligation artifacts found in and removed from the read mapping.
- QC_report: Contains quality information such as sequence length, GC content and quality, to help detect any sequencing bias in the sample reads.
- structural_variants_report: Provides an overview of any potential structural variants found in the sample.
- mapping_report: Summarizes the number of mapped and unmapped reads, as well as providing coverage statistics for the SARS-CoV-2 reference genome.
- coverage_report: Gives an overview of coverage in the targeted regions e.g. minimum coverage in % of target regions and number of targets passing the coverage threshold of 30x. For the Ion AmpliSeq protocol, "target regions" refers to the targeted amplicons. For the other workflows Target region is defined by the full length genome. The coverage report is helpful for identifying regions where coverage is insufficient to reliably call variants. Variants present in regions with insufficient coverage is not called although the read mapping shows evidence of the variant being present in the reads. The report can help understand such results and be used for adjusting coverage requirements if deemed necessary.
- variant_report: Contains information about the estimated error model for the variant calls.
- variant_track_statistics_report: Contains information about statistics on variants classified by types.
- human_control_genes_mapping_report (Ion AmpliSeq SARS-CoV-2 Low Frequency and Shared Variants workflow only): Contains mapping information for the 5 human expression gene controls.
Tracks
The following tracks are produced:
- breakpoints: Contains a row for each potential breakpoint with information on region, p-value, mapping information and number of reads supporting breakpoint.
- InDels_track: The InDels used as guidance variants for the local realignment. Note, in some cases it might be necessary to create the consensus sequence using the coordinates of the full insertion from the InDels track. This can happen when no reads span the entire region.
- realigned_regions: Contains a row for each region in which the mapping was improved following local realignment.
- read_mapping: The read mapping track after local realignment, trimming and removal of marginal reads.
- coverage_below_30: Shows regions that failed to meet the coverage threshold of 30x and were therefore not used when calling variants nor used towards building the consensus sequence.
- amino_acid_track: Shows the consequences of the variants at the amino acid level in the context of the original amino acid sequence. A variant introducing a stop mutation is displayed in red.
- coverage: Coverage per position across the SARS-CoV-2 reference.
- variants_above_50_frequency: A list of variants that passed the >=50% frequency quality filter. There is strong evidence these variants are present in the sample.
Genome View
The Genome View track list contains the following collection of tracks:
- The reference SARS-CoV-2 genome MN908947.3
- The reference SARS-CoV-2 CDS regions
- Amino acid changes track produced using unfiltered variant calls
- Coverage graph
- Read mapping after local realignment, trimming and removal of marginal reads
- The reference SARS-CoV-2 gene regions
- Low coverage regions (<30x coverage)
- Variants with >=50% frequency that passed the quality filter
An example is shown in figure 22.8.
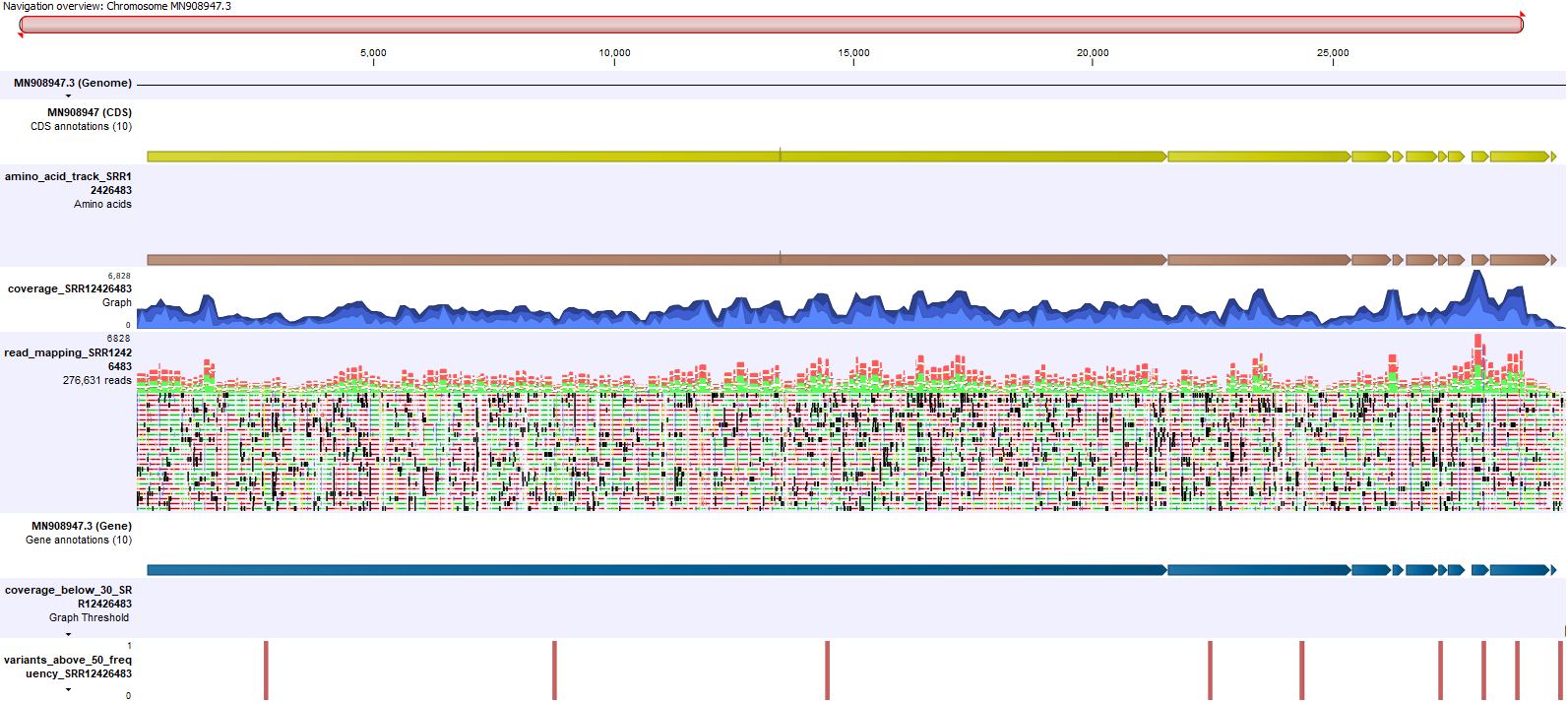
Figure 22.8: An example of the Genome View track list created for each sample
Supplemental
The following results are placed in a folder called Supplemental:
- unmapped reads: Reads that did not map to the SARS-CoV-2 reference or the human control genes when using the Ion AmpliSeq SARS-CoV-2 Low Frequency and Shared Variants workflow.
- unfiltered_variant_track: The variant track generated by Low Frequency Variant Detection before any further filtering was carried out.
- low_frequency_variants: A track containing all variants passing the quality filter with a frequency (Optional value, Default >=10% or 20%).