Create UMI Reads from Reads
Create UMI Reads from Reads generates a single consensus read, called a UMI read, from the reads in the input sequence list(s) that share the same or similar UMI and have similar sequences. The tool can be used to process single end reads and paired end reads, including reads generated by duplex sequencing methods. Reads must be preprocessed by Remove and Annotate with Unique Molecular Index. It is recommended that the reads are also trimmed for adapters and homopolymers.
The tool outputs a sequence list containing consensus UMI reads. Optional outputs include a QC Report and a list of discarded reads.
When present, read group information from the input sequence lists is preserved in the output UMI reads, provided that the input lists share the same read group. For samples sequenced on multiple lanes this requires that the reads from the different lanes are joined during import.
The algorithm is loosely inspired by Mash (Ondov 2016), Linclust (Steinegger 2017) and Calib (Orabi 2017) and involves clustering similar reads, merging reads in a cluster with the same UMI, and then filtering the merged reads. These steps are described in detail below:
Grouping reads
The tool makes extensive use of the minHash concept for Locality-Sensitive Hashing (LSH) to find clusters of similar reads. Briefly, all k-mers of each read are hashed with a number of hash functions, and for each of these hash functions the lowest hash value over all the k-mers is recorded. Two reads that share a lowest hash value are likely to share a k-mer and are linked together. In practice, a single hash function is not sufficiently specific to link related reads, and so sets of hash functions are used. Reads are only linked if each hash function in the set has the same lowest value for the two reads. The linked reads can be thought of as forming a graph, where each read is a node, and edges are made between the linked reads. Clusters of reads form disconnected subgraphs i.e. they are only linked to each other.
The tool uses LSH in three rounds:
1) In the first round reads are 'coarse clustered', so that it is very likely that similar reads are in the same cluster. The clusters are 'coarse' because it is also likely that unrelated reads are in the same cluster. The UMI is not used during coarse clustering.
2) In the second round each coarse cluster is 'fine clustered' by applying LSH again, but using different hash settings in order to get a more precise clustering. Reads in the resulting clusters are 'merged' if they have the same UMI.
3) In the third round all reads for a coarse cluster (merged or not) are again 'fine clustered' by LSH. Each disconnected subgraph is then pruned to only keep links between reads where (i) the UMIs differ by one mismatch, and (ii) either one of the reads is not merged, or the `weight' of one of the reads is more than twice the weight of the other (this is the directional method in Smith 2017). The `weight' of a merged read is the number of raw reads that were used to form the merged read.
Merging reads
The merging of similar UMI reads is performed using Multiple Sequence Alignment (MSA) based on the SPOA C++ library (https://github.com/rvaser/spoa). In general terms, the POA method creates a graph whose nodes represent the bases of the first sequence, and aligns the second sequence to the graph. Aligned bases of the first/second sequence are then merged in the graph. Every new sequence is aligned to the graph (using a modified version of the usual dynamic programming algorithms) and merged into the graph. As bases are merged into the graph, the edges between the nodes are updated to reflect their `weight', which corresponds to the number of times they occurred in multiple sequences. Finally, when all sequences are aligned, the graph is traversed to find the `heaviest' path, which is the consensus sequence.
When sequences are aligned, mismatching nodes are connected by a special type of edge. Using these edges, it is possible to find all mismatches of a base in the consensus read. With this information in hand, the quality consensus is calculated using a method similar to [Hiatt et al., 2013], which is also used in the other UMI tools. Every node keeps a match and a mismatch quality value (because it is not possible to know at the time of graph construction if it will be a match or not). Every time a new sequence is added and a new node is merged the quality values are updated using the quality scores of the new sequence. After calculating the consensus, the values in each node reflect the improved quality scores due to multiple sequences calling the same base. The quality consensus is obtained by processing the match and a mismatch quality value accordingly as described in [Hiatt et al., 2013].
Duplex UMI
Reads produced by the TSO (TruSight Oncology 500, Illumina) duplex sequencing protocol can be processed to create a duplex consensus if reads from both strands are available. To create this consensus, reads probably originating from the same molecule and strand are grouped based on their similarity, UMI sequence and strand information. Single-strand consensus sequences are generated from these groups. Then, the consensus sequences of the opposing strands are combined to generate the duplex consensus sequences.
Background information:
A protocol is considered duplex if the unique molecular barcode(s) attached to the target molecules include enough information to determine if a read originates from the same or from the opposite strand of the same target molecule. For the TSO protocol, the dual stranded reads have different barcodes attached to each end of the fragments, resulting in each read having two different barcodes named alpha and beta. Reads with the same orientation have identical alpha and beta tags, whereas reverse complements of the reads have alpha reads matching beta reads and vice versa (see Remove and Annotate with Unique Molecular Index for further information).
Running the tool
Create UMI Reads from Reads is available under the Tools menu at:
Tools | Biomedical Genomics Analysis (![]() ) | UMI Tools (
) | UMI Tools (![]() ) | Create UMI Reads from Reads (
) | Create UMI Reads from Reads (![]() )
)
In the first dialog, select the sequence list containing the reads (figure 4.6).
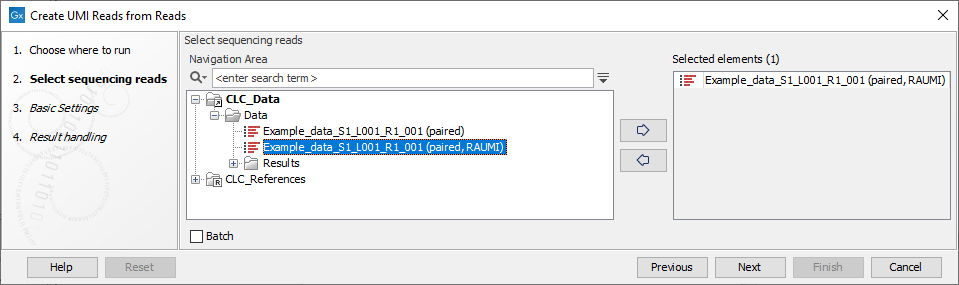
Figure 4.6: Select the sequencing reads by double-clicking on the file name or by clicking once on the file name and then on the arrow pointing to the right hand side.
The next dialog allows you to configure basic settings for this tool, as shown in figure 4.7 and described below.
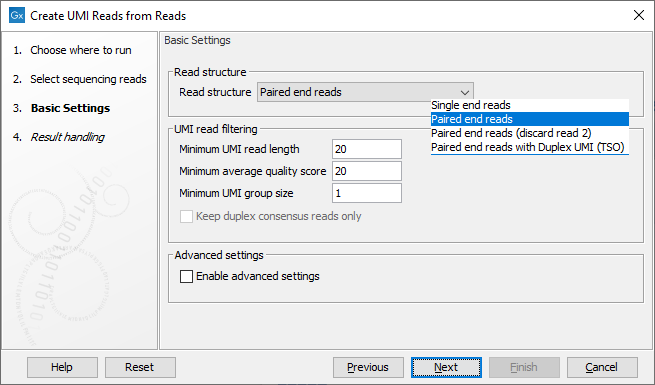
Figure 4.7: Basic settings of the Create UMI Reads from Reads tool.
- Read structure The read type and how they should be processed. The options are:
- Single end reads When selected, each step of the algorithm described above is performed on each read.
- Paired end reads When selected, each step of the algorithm described above is performed on read 1 (R1) and read 2 (R2) separately. During the hashing step, specific hash nodes are created only for R1. The purpose is to keep track of the UMI barcode and the LSH hashes. To link R1 and R2 a paired UMI reads object is created. In the final part of merging, reads are only combined in paired reads if R1 and R2 are similar. Strand specificity is not considered when this option is selected.
- Paired end reads (discard read 2) Only the R1 member of a pair is processed. Discarding R2 is useful when it does not contain any biological information.
- Paired end reads with Duplex UMI (TSO) Appropriate for reads generated using the TSO protocol. A duplex consensus will be generated. Here, both reads are hashed and added to the list for grouping. R1 hashes together with "alpha + beta" UMI, and R2 hashes together with "beta + alpha" UMI. In this way, reads from both strands end up in the same "coarse" groups. In the final part of merging, reads are only combined in paired reads if R1 and R2 are similar, the same as for the "Paired end reads" case, described above.
- Minimum UMI read length: UMI reads shorter than this value will be discarded.
- Minimum average quality score: UMI reads will be discarded, if their average Q-score is lower than the value specified here.
- Minimum group size: The tool will only create a UMI read if the number of reads in the UMI group is at least the size specified here.
- Keep duplex consensus reads only: When selected, only UMI reads built from reads representing both strands are retained. This is only relevant when a duplex UMI option has been selected for the "Read structure" setting.
- Enable advanced settings: When enabled, the advanced settings, available in the next wizard step, can be configured.
The next wizard step lists the advanced settings, which are organized in three categories: "Consensus options", "Coarse grouping" options, used for the coarse clustering step, and "Fine grouping" options, using for the fine clustering step (figure 4.8).
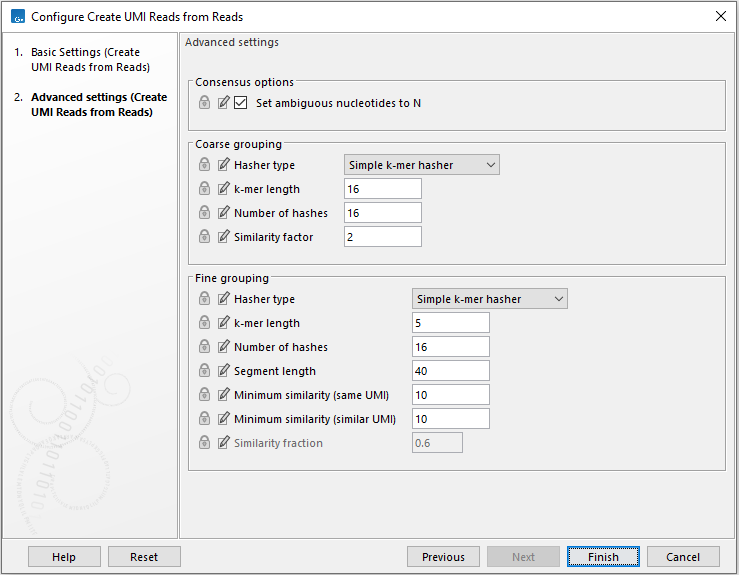
Figure 4.8: Advanced settings of the Create UMI Reads from Reads tool.
As both clustering steps use the same algorithm, their parameters are similar.
- Hasher type:
- Simple k-mer hasher: Hashes are computed for every k-mer of a read.
- Spaced seed hasher: Hashes are computed over multiple subsets of positions within the k-mer. For example, a spaced seed hasher of length 5 might make shorter 3-mers of positions 1,2,5 and 1,3,5.
- Rolling min k-mer hasher: Hashes are the minimum k-mer hashes within k-mer length of each other for all read bases. This hasher can group reads that do not map to the same position, because the overlapping parts of the reads will have the same hashes.
Note that for single primer extension protocols, reads representing the same DNA fragment can originate from different primers. This can happen if primers in the same direction are located near each other, making it possible for a downstream primer to amplify a PCR product generated from an upstream primer.
These types of reads will not be grouped using the Simple k-mer hasher option, but they will be grouped when using the Rolling min k-mer hasher option.
When reads originating from different primers are not grouped, this can lead to too high coverage. When there are few reads per UMI, this effect is expected to be negligible.
When reads originating from different primers are grouped, primer sequence from reads starting downstream of another read is incorporated in the consensus UMI read. Because the primer sequence is always the reference sequence, this can lead to correction of variants to reference sequence when the consensus is calculated. This can cause variant frequencies to become skewed, because a subset of UMI reads will have been corrected from variant to reference. Fusion detection and RNA expression results are unlikely to be affected by skewed variant frequencies. When there are few reads per UMI, this effect is expected to be negligible.
- K-mer length:
- Simple k-mer hasher: A length in the range 2-32. Shorter k-mers lead to coarser clusters.
- Spaced seed hasher: A length of 5, 8, 12, or 16. Shorter k-mers lead to coarser clusters.
- Rolling min k-mer hasher: A length below 27. Shorter k-mers lead to coarser clusters.
- Number of hashes: the total number of hash functions applied to each k-mer of the read
- Similarity factor / Minimum similarity (same UMI) / Minimum similarity (similar UMI): hash functions are divided into groups of this size. For two reads to be linked, all the hash functions in the group must have the same minimum hash values for the two reads. Therefore the number of hashes must be an exact multiple of this number. The higher this value, the finer the clusters.
- Similarity fraction: Minimum fraction of equal hashes between two reads required to group them. This is used by the rolling min k-mer hasher to handle reads with different number of hashes.
The remaining parameters are:
- Segment length: Only compute hashes over this number of bases at the start of a read. By only clustering on the start of the read, we reduce the chance of merging reads with different start positions (and which therefore likely come from different fragments) into a consensus UMI read.
- Set ambiguous nucleotide to N (checked by default): This option determines whether to attempt to error correct UMI reads, or to replace conflict positions with 'N' to indicate that there is uncertainty about the true nucleotide. Error correction works by majority vote, and is useful when the predominant source of error is expected to be sequencing error. Replacing conflict positions with 'N' is more conservative, effectively discarding a position. This can be desired when errors are introduced by library preparation.
Click Next to Open or Save the sequence list of merged UMI reads.
It is also possible to generate a report that will indicate how many reads were ignored and the reason why they were not included in a UMI read.
The report also contains group size (see
UMI group sizes)
and quality score statistics useful for QC.