Running multimodal workflows in batch using metadata
In order to run the workflow in batch, metadata must be provided to describe which DNA and RNA reads belong together.
See https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Running_workflows_in_batch_mode.html for details on batch analysis, https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Metadata.html for general information about metadata, and https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Importing_metadata.html for how to import a metadata table.
Metadata can easily be supplied from an Excel spreadsheet or CSV file. A minimal spreadsheet should contain the start of the file names and a column containing the sample information:
| Fastq | Sample |
| DNA-S1 | S1 |
| RNA-S1 | S1 |
| DNA-S2 | S2 |
| RNA-S2 | S2 |
Start the workflow normally, but remember to tick batch twice - once when selecting DNA reads (figure 14.2), and again when selecting RNA reads (figure 14.3).
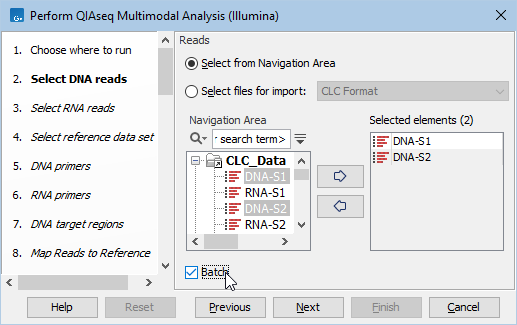
Figure 14.2: Both DNA samples are selected at the same time. They will be grouped into batches in a later step.
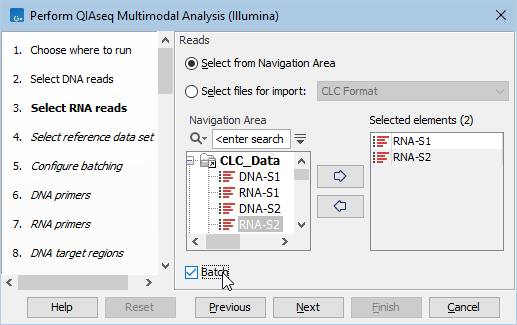
Figure 14.3: Both RNA samples are selected at the same time. They will be grouped into batches in a later step.
Metadata can be chosen in the 'Configure batching' dialog (figure 14.4).
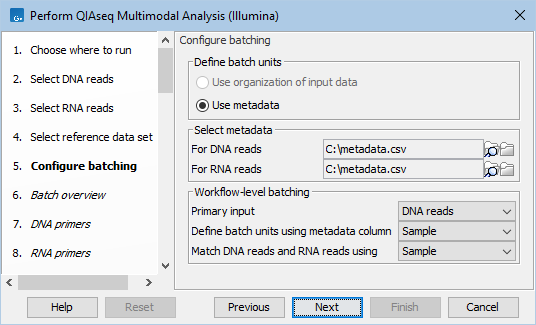
Figure 14.4: Configuration of batch units based on metadata. Each batch unit is named after the DNA file name. DNA and RNA reads are grouped together if they share a value in the 'Sample' column.
The next dialog will show how the batching will be performed (figure 14.5).
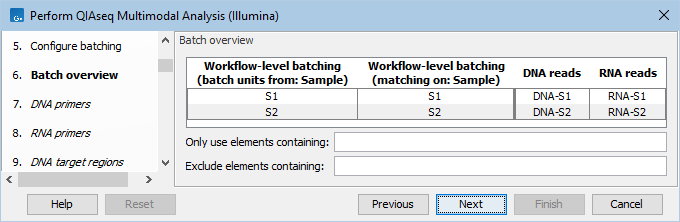
Figure 14.5: Overview of the batch units. Each batch unit is named after the DNA file name. DNA and RNA reads are grouped together if they share a value in the 'Sample' column.