Microhaplotype Caller (beta)
The Microhaplotype Caller (beta) identifies sample alleles at variant loci as well as specified marker loci, uses phasing information available in the mapped reads to infer haplotypes, and produces a Genotype track with all detected alleles, including, for inspection, those not called due to applied filters.
The Microhaplotype Caller (beta) takes advantage of the same probabilistic approach as the Low Frequency Variant Detection tool, it is therefore suitable for analysis of mixed tissue samples in which low frequent variants are likely to be present, as well as for samples for which the ploidy is unknown or not well defined. For method details, see: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=_Low_Frequency_Variant_Detection_tool_Models_methods.html
Key features distinguishing the Microhaplotype Caller (beta) method:
- The long variant detection responsible for calling MNVs, replacements, and indels in the Low Frequency Variant Detection tool, has been adapted to also call longer more general haplotypes. This enables high resolution allele detection, so multiple haplotype alleles may be distinguished per allele variant and reported with detailed annotations.
- Detected alleles are reported in a locus based representation, making genotypes readily available, e.g. for VCF export.
- When filters are applied, the affected alleles are retained for easy user inspection.
- Forced variant loci can be specified to allow detection of alleles at any site, also homozygous reference loci.
The current version of the Microhaplotype Caller (beta) has certain limitations:
- If phasing regions become too large the CLC Workbench may run out of memory. Caution must therefore be taken when increasing the 'Maximum phasing distance' parameter from the default value (see below).
- There is not yet any special homopolymer handling, so length variation artifacts must be considered when analysing loci around longer homopolymers.
Toolbox | Biomedical Genomics Analysis (![]() ) | Haplotype Calling (beta) (
) | Haplotype Calling (beta) (![]() ) | Microhaplotype Caller (beta) (
) | Microhaplotype Caller (beta) (![]() )
)
The Microhaplotype Caller (beta) has three options to select information available in the output (figure 10.5):
- Forced loci: Report alleles for the specified loci, including homozygous reference alleles. Alleles detected at other variant loci will still be reported. Either a Genotype track or an Annotation track can be selected.
- Maximum phasing distance: The maximum positional distance from one locus to nearest neighbour locus, to include them in the same phasing region. For example, to ensure alleles are phased within a codon, set this parameter to 2.
WARNING: Setting this parameter high may lead to performance issues. This parameter must be kept around its default value when a high density of variant loci is expected to be detected, for example due to a combination of high read coverage, poor read quality, and high sensitivity detection parameters.
- Restrict calling to target regions: Variant detection will only be performed in the specified regions.
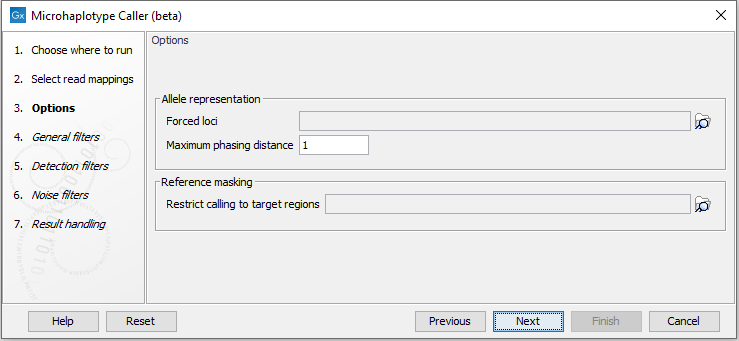
Figure 10.5: The Microhaplotype Caller options.
General filters
There are three main properties by which the output can be filtered, and each of these have a detection and a call threshold (figure 10.6). The detection threshold specifies if elements are reported or not, whereas the call threshold specifies whether an element is marked with a filter to be disregarded as noise. Elements with applied filters are however still reported for inspection, which can be useful for evaluating borderline calls.
- Minimum count (detection): Alleles observed in less than this number of reads will not be detected and can therefore not be reported.
- Minimum count (call): Alleles observed in less than this number of reads will not be called though will be reported as filtered if detected.
- Minimum allele fraction (detection): Alleles observed at less than this fraction will not be detected and can therefore not be reported.
- Minimum allele fraction (call): Alleles observed at less than this fraction will not be called though will be reported as filtered if detected.
- Minimum haplotype quality (detection): Haplotypes with lower quality will not be detected and can therefore not be reported.
- Minimum haplotype quality (call): Haplotypes with lower quality will not be called though will be reported as filtered if detected.
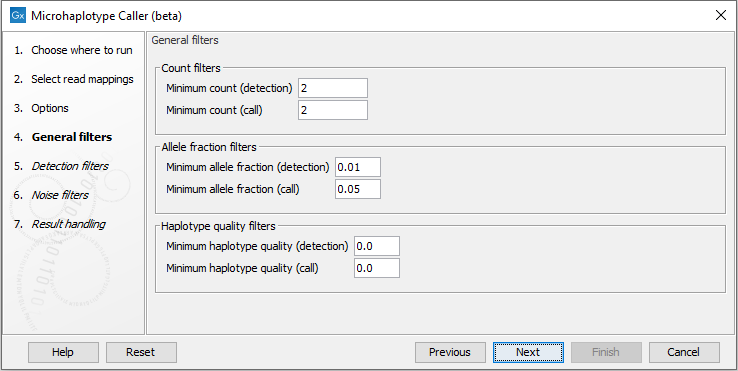
Figure 10.6: The Microhaplotype Caller general filters.
Detection filters
The detection filters affect the sensitivity of the Microhaplotype Caller (beta) (figure 10.7):- Required Significance: this parameter determines the cut-off value for the statistical test for the variant not being due to sequencing errors. Only variants that are at least this significant will be called. The lower you set this cut-off, the fewer variants will be called.
- Ignore broken pairs: When enabled, reads from broken pairs are ignored. Broken pairs may arise for a number of reasons, one being erroneous mapping of the reads. In general, variants based on broken pair reads are likely to be less reliable, so ignoring them may reduce the number of spurious variants called. However, broken pairs may also arise for biological reasons (e.g. due to structural variants) and if they are ignored some true variants may go undetected. Please note that ignored broken pair reads will not be considered for any non-specific match filters.
- Non-specific match filter: Non-specific matches are likely to come from repeat region whose exact mapping location is uncertain. In general, variants based on non-specific matches are likely to be less reliable. However, as there are regions in the genome that are entirely perfect repeats, ignoring non-specific matches may have the effect that true variants go undetected in these regions.
There are three options for specifying to which 'extent' the non-specific matches should be ignored:
- 'No': they are not ignored.
- 'Reads': they are ignored.
- 'Region': when this option is chosen no variants are called in regions covered by at least one non-specific match. In this case, the minimum length of reads that are allowed to trigger this effect has to be stated, as really short reads will usually be non-specific even if they do not stem from repeat regions.
- Base quality filter: The base quality filter can be used to ignore the reads whose nucleotide at the potential variant position is of dubious quality. This is assessed by considering the quality of the nucleotides in the region around the nucleotide position. There are three parameters to determine the base quality filter:
- Neighborhood radius: This parameter determines the region size. For example if a neighborhood radius of five is used, a nucleotide will be evaluated based on the nucleotides that are 5 positions upstream and 5 positions downstream of the examined site, for a total of 11 nucleotides. Note that, near the end of the reads, eleven nucleotides will still be considered by offsetting the region relative to the nucleotide in question.
- Minimum central quality: Reads whose central base has a quality below the specified value will be ignored. This parameter does not apply to deletions since there is no 'central base' in these cases.
- Minimum neighborhood quality: Reads for which the minimum quality of the bases is below the specified value will be ignored.
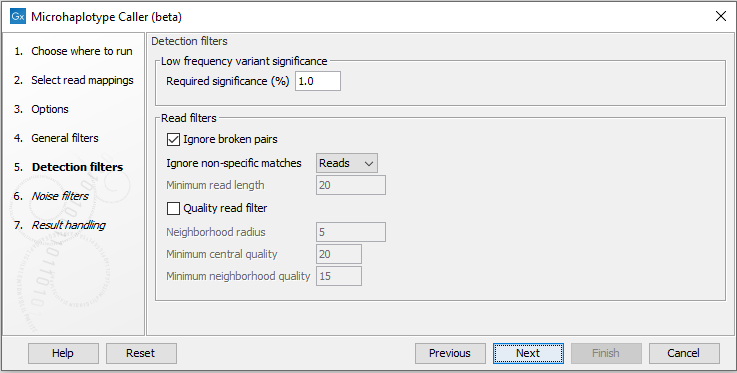
Figure 10.7: The Microhaplotype Caller detection filters.
Noise filters
These filter thresholds specify when a filter is applied to a genome element for it to be disregarded as noise (figure 10.8):- Minimum coverage: Only variants in regions covered by at least this many reads are called.
- Maximum coverage: All positions with coverage above this value will be ignored when inspecting the read mapping for variants. The option is highly useful in cases where you have a read mapping which has areas of extremely high coverage as are areas around centromeres for example.
- Minimum average base quality: Alleles that have an average base quality below this threshold are disregarded as noise.
- Read direction filter: The read direction filter removes alleles that are almost exclusively present in either forward or reverse reads. For many sequencing protocols such alleles are most likely to be the result of amplification induced errors. Note, however, that the filter is NOT suitable for amplicon data, as for this you will not expect coverage of both forward and reverse reads. The filter has a single parameter:
- Direction frequency: A filter is applied to alleles that are not supported by at least this frequency of reads from each direction.
- Relative read direction filter: The relative read direction filter attempts to do the same thing as the 'Read direction filter', but does this in a statistical, rather than absolute, sense: it tests whether the distribution among forward and reverse reads of the variant carrying reads is different from that of the total set of reads covering the site. The statistical, rather than absolute, approach makes the filter less stringent. The filter has one parameter:
- Significance: A filter is applied to alleles whose read direction distribution is significantly different from the expected with a test at this level. The lower you set the significance cut-off, the fewer alleles will be filtered out.
- Read position filter: The read position filter is a filter that attempts to remove systematic errors in a similar fashion as the 'Read direction filter', but that is also suitable for hybridization-based data. It removes alleles that are located differently in the reads carrying it than would be expected given the general location of the reads covering the variant site. This is done by categorizing each sequenced nucleotide (or gap) according to the mapping direction of the read and also where in the read the nucleotide is found; each read is divided in five parts along its length and the part number of the nucleotide is recorded. This gives a total of ten categories for each sequenced nucleotide and a given site will have a distribution between these ten categories for the reads covering the site. If a distinct allele is present in the site, you would expect the allele nucleotides to follow the same distribution. The read position filter carries out a test for whether the read position distribution of the allele carrying reads is different from that of the total set of reads covering the site. The filter has one parameter:
- Significance: A filter is applied to alleles whose read position distribution is significantly different from the expected with a test at this level. The lower you set the significance cut-off, the fewer alleles will be filtered out.
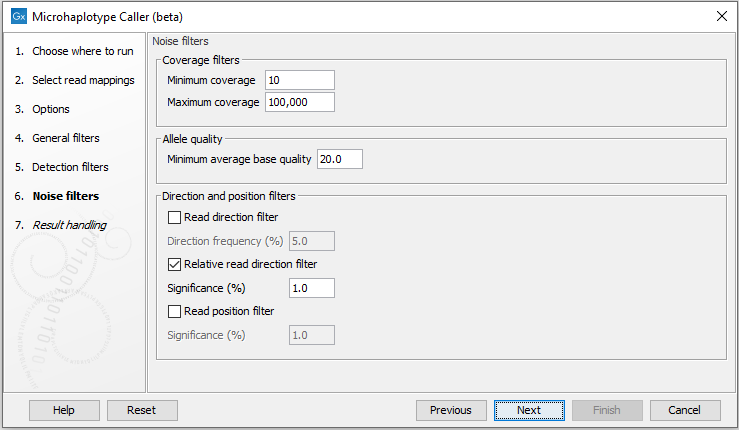
Figure 10.8: The Microhaplotype Caller noise filters.
Subsections