Identify QIAseq Exome Germline Variants
The Identify QIAseq Exome Germline Variants template workflow is designed to call germline variants from data generated with QIAseq Human Exome kits.
The first steps of these workflow involve trimming off any remaining PCR adapters. This is followed by mapping the trimmed reads to the human reference sequence. The Structural Variant Caller then generates a guidance track that is used in the Local Realignment tool to improve the mapping. The improved mapping is then input to the Fixed Ploidy Variant Detection tool. The resulting variants are filtered to remove those located outside defined target regions. Remaining variants are then annotated with information such as the relation to repeat/homopolymer regions or gene elements. Finally, a series of filtering steps removes variants likely to be artifacts. The retained variants are output, along with reports and other associated results.
The workflow can be found at:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | QIAseq DNA workflows (
) | QIAseq DNA workflows (![]() ) | Identify QIAseq Exome Germline Variants (
) | Identify QIAseq Exome Germline Variants (![]() )
)
If you are connected to a CLC Server via the CLC Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the "Select Reads" wizard step, specify the data to be analyzed (figure 13.53). This workflow expects sequencing reads as input. When selecting data from the Navigation Area, these would normally be supplied as sequence lists.
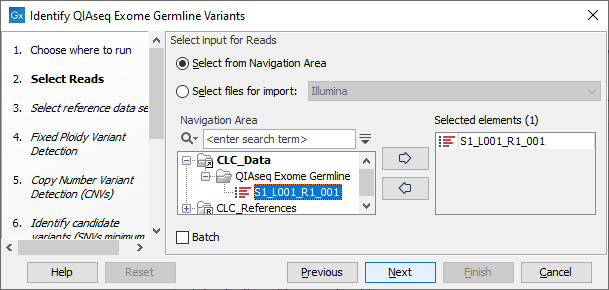
Figure 13.53: Select the data to analyze.
The following dialog helps you set up the relevant Reference Data Set. If you have not downloaded the Reference Data Set yet, the dialog will suggest the relevant data set and offer the opportunity to download it using the Download to Workbench button. We recommend that the default QIAseq Exome hg38 reference data set is selected for use, see figure 13.54.
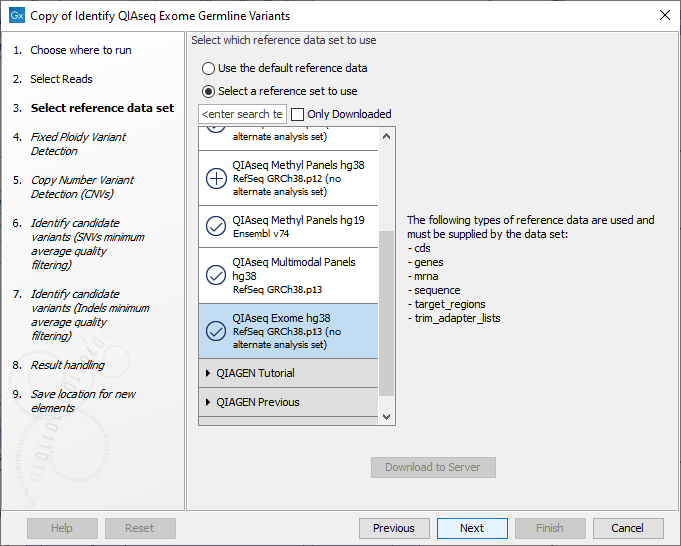
Figure 13.54: The relevant Reference Data Set is highlighted. To the right, the types of reference data needed by the workflow are listed.
Note that if you wish to Cancel or Resume the Download, you can close the template workflow and open the Reference Data Manager where the Cancel, Pause and Resume buttons are available.
If the Reference Data Set was previously downloaded, the option "Use the default reference data" is available and will ensure the relevant data set is used. You can always check the "Select a reference set to use" option to be able to specify another Reference Data Set than the one suggested.
In the Map Reads to Reference dialog, it is possible to configure masking. A custom masking track can be used, but by default, the masking track is set to GenomeReferenceConsortium_masking_hg38_no_alt_analysis_set, containing the regions defined by the Genome Reference Consortium, which serve primarily to remove false duplications, including one affecting the gene U2AF1. Changing the masking mode from "No masking" to "Exclude annotated" excludes these regions.
In the Fixed Ploidy Variant Detection wizard step, the variant detection options can be configured (figure 13.55). These are:
- Minimum coverage: Only variants in regions covered by at least this many reads are called.
- Minimum count: Only variants that are present in at least this many reads are called.
- Minimum frequency: Only variants that are present at least at the specified frequency (calculated as 'count'/'coverage') are called.
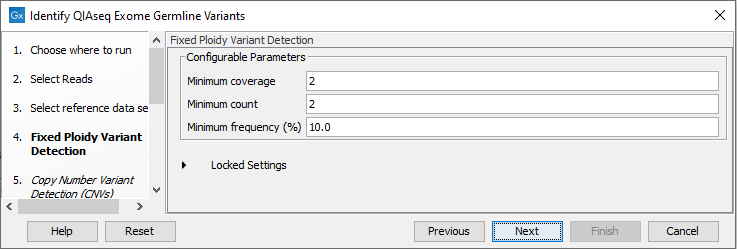
Figure 13.55: Specify the parameters for the Fixed Ploidy Variant Detection tool.
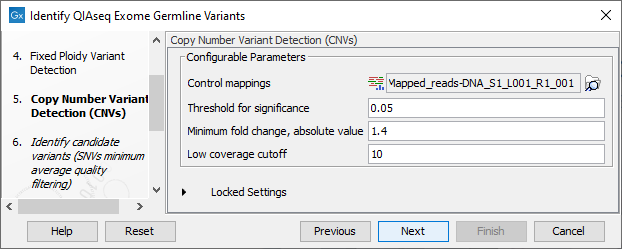
Figure 13.56: Specify the parameters for the Copy Number Variant Detection tool.
The dialog for Copy Number Variant Detection allows you to specify a control mapping against which the coverage pattern in your sample will be compared in order to call CNVs. If you do not specify a control mapping the Copy Number Variation analysis will not be carried out (figure 13.56).
Please note that if you want the copy number variation analysis to be done, it is important that the control mapping supplied is a meaningful control for the sample being analyzed. Mapping of control samples for the CNV analysis can be done using the workflow described in Create QIAseq Exome CNV Control Mapping.
A meaningful control must satisfy two conditions: (1) It must have a copy number status that is meaningful to compare against. For panels with targets on the X and Y chromosomes, the control and sample should be matched for gender. (2) The control read mapping must result from the same type of processing that will be applied to the sample.
In the next two dialogs individual filtering settings can be specified for SNVs and indels.
In the final wizard step, choose to Save the results of the workflow and specify a location in the Navigation Area before clicking Finish.
Launching using the QIAseq Panel Analysis Assistant
The workflow is also available in the QIAseq Panel Analysis Assistant under Exome.
Subsections