Roche 454
Choosing the Roche 454 import will open the dialog shown in figure 6.4.
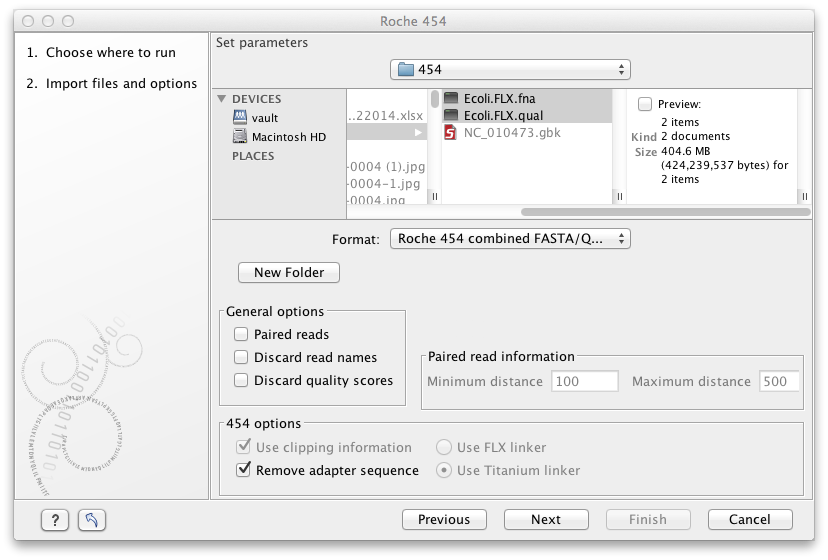
Figure 6.4: Importing data from Roche 454.
We support import of two kinds of data from 454 GS FLX systems:
- Flowgram files (.sff ) which contain both sequence data and quality scores amongst others. However, the flowgram information is currently not used by CLC Genomics Workbench. There is an extra option to make use of clipping information (this will remove parts of the sequence as specified in the .sff file).
- Fasta/qual files:
- 454 FASTA files (.fna ) which contain the sequence data.
- Quality files (.qual ) which contain the quality scores.
For all formats, compressed data in gzip format is also supported (.gz).
The General options to the left are:
- Paired reads. The paired protocol for 454 entails that the forward and reverse reads are separated by a linker sequence. During import of paired data, the linker sequence is removed and the forward and reverse reads are separated and put into the same sequence list (their status as forward and reverse reads is preserved). You can change the linker sequence in the Preferences (in the Edit menu) under Data. Since the linker for the FLX and Titanium versions are different, you can choose the appropriate protocol during import, and in the preferences you can supply a linker for both platforms (see figure 6.5. Note that since the FLX linker is palindromic, it will only be searched on the plus strand, whereas the Titanium linker will be found on both strands. Some of the sequences may not have the linker in the middle of the sequence, and in that case the partial linker sequence is still removed, and the single read is put into a separate sequence list. Thus when you import 454 paired data, you may end up with two sequence lists: one for paired reads and one for single reads. Note that for de novo assembly projects, only the paired list should be used since the single reads list may contain reads where there is still a linker sequence present but only partially due to sequencing errors. Read more about handling paired data.
- Discard read names. For high-throughput sequencing data, the naming of the individual reads is often irrelevant given the huge amount of reads. This option allows you to discard this option to save disk space.
- Discard quality scores. Quality scores are visualized in the mapping view and they are used for SNP detection. If this is not relevant for your work, you can choose to Discard quality scores. One of the benefits from discarding quality scores is that you will gain a lot in terms of reduced disk space usage and memory consumption. If you have selected the fna/qual option and choose to discard quality scores, you do not need to select a .qual file.
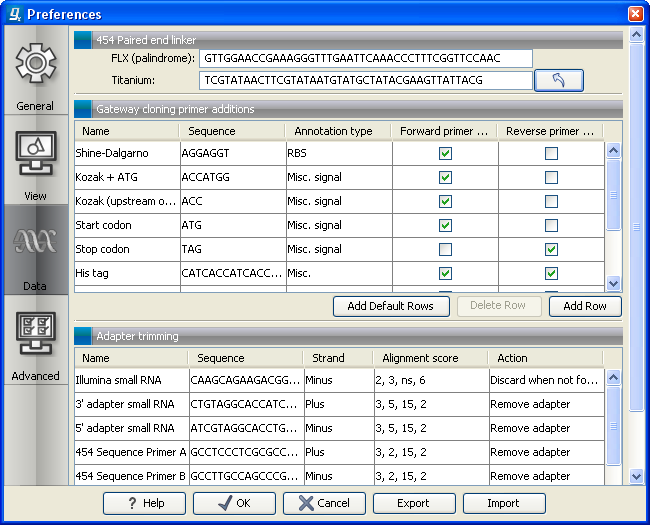
Figure 6.5: Specifying linkers for 454 import.
Note! During import, partial adapter sequences are removed (TCAG and ATGC ), and if the full sequencing adapters GCCTTGCCAGCCCGCTCAG , GCCTCCCTCGCGCCATCAG or their reverse complements are found, they are also removed (including tailing Ns). If you do not wish to remove the adapter sequences (e.g. if they have already been removed by other software), please uncheck the Remove adapter sequence option.
Click Next to adjust how to handle the results. We recommend choosing Save in order to save the results directly to a folder, since you probably want to save anyway before proceeding with your analysis. There is an option to put the import data into a separate folder. This can be handy for better organizing subsequent analysis results and for batch processing.