Create virtual tag list
Before annotating the tag sample (The virtual tag list can be saved and used to annotate experiments made from tag-based expression samples as shown in Annotate tag samples or experiment.
To create the list:
Toolbox | Transcriptomics Analysis (![]() ) | Expression Profiling by Tags (
) | Expression Profiling by Tags (![]() ) | Create Virtual Tag List (
) | Create Virtual Tag List (![]() )
)
This will open a dialog where you select one or more annotated genomic sequences or a list of ESTs. Click Next when the sequences are listed in the right-hand side of the dialog.
This dialog is where you specify the basis for extracting the virtual tags (see figure 26.39).
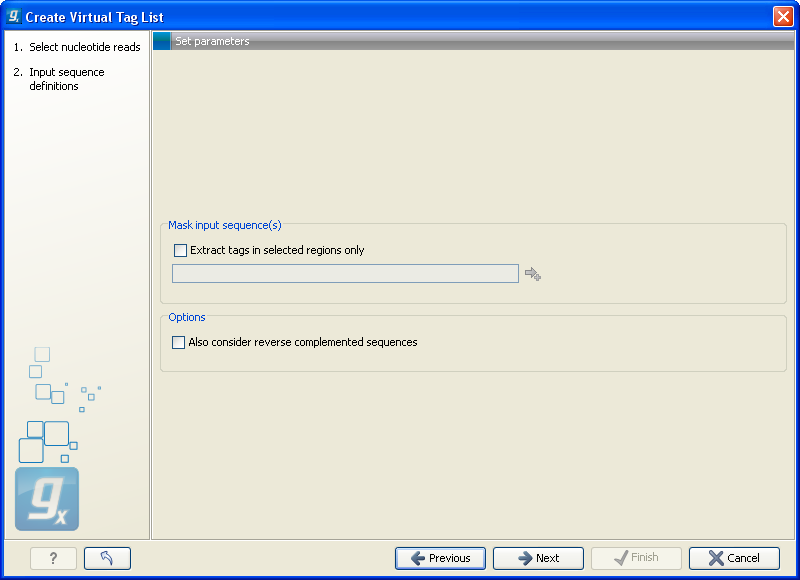
Figure 26.39: The basis for the extraction of reads.
At the top you can choose to extract tags based on annotations on your sequences by checking the Extract tags in selected areas only option. This option is applicable if you are using annotated genomes (e.g. Refseq genomes). Click the small button (![]() ) to the right to display a dialog showing all the annotation types in your sequences. Select the annotation type representing your transcripts (usually mRNA or Gene). The sequence fragments covered by the selected annotations will then be extracted from the genomic sequence and used as basis for creating the virtual tag list.
) to the right to display a dialog showing all the annotation types in your sequences. Select the annotation type representing your transcripts (usually mRNA or Gene). The sequence fragments covered by the selected annotations will then be extracted from the genomic sequence and used as basis for creating the virtual tag list.
If you use a sequence list where each sequence represents your transcript (e.g. an EST library), you should not check the Extract tags in selected areas only option.
Below, you can choose to include the reverse complement for creating virtual tags. This is mainly used if there is uncertainty about the orientation of sequences in an EST library.
Clicking Next allows you to specify enzymes and tag length as shown in figure 26.40.
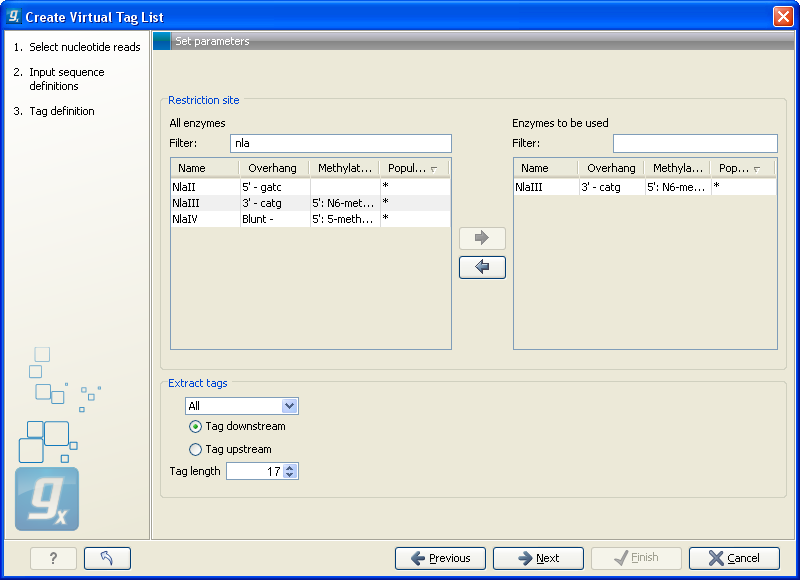
Figure 26.40: Defining restriction enzyme and tag length.
At the top, find the enzyme used to define your tag and double-click to add it to the panel on the right (as it has been done with NlaIII in figure 26.40). You can use the filter text box so search for the enzyme name.
Below, there are further options for the tag extraction:
- Extract tags
- When extracting the virtual tags, you have to decide how to handle the situation where one transcript has several cut sites. In that case there would be several potential tags. Most tag profiling protocols extract the 3'-most tag (as shown in the introduction in figure 26.34), so that would be one way of defining the tags in the virtual tag list. However, due to non-specific cleavage, new alternative splicing or alternative polyadenylation [t Hoen et al., 2008], tags produced from internal cut sites of the transcript are also quite frequent. This means that it is often not enough to consider the 3'-most restriction site only. The list lets you select either All, External 3' which is the 3'-most tag or External 5' which is the 5' most tag (used by some protocols, for example CAGE - cap analysis of gene expression - see [Maeda et al., 2008]). The result of the analysis displays whether the tag is found at the 3' end or if it is an internal tag (see more below).
- Tag downstream/upstream
- When the cut site is found, you can specify whether the tag is then found downstream or upstream of the site. In figure 26.34, the tag is found downstream.
- Tag length
- The length of the tag to be extracted. This should correspond to the sequence length defined in figure 26.35.
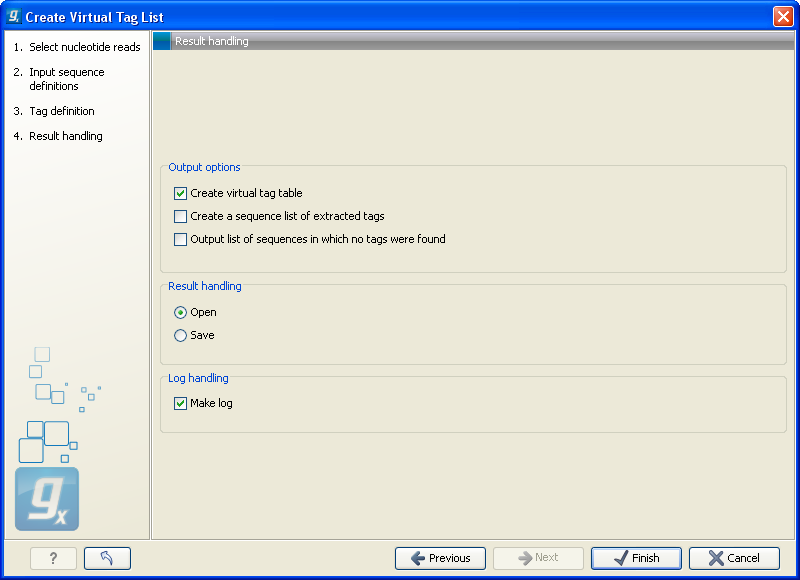
The output options are:
- Create virtual tag table
- This is the primary result listing all the virtual tags. The table is explained in detail below.
- Create a sequence list of extracted tags
- All the extracted tags can be represented in a raw sequence list with no additional information except the name of the transcript. You can e.g. Export (
 ) this list to a fasta file.
) this list to a fasta file.
- Output list of sequences in which no tags were found
- The transcripts that do not have a cut site or where the cut site is so close to the end that no tag could be extracted are presented in this list. The list can be used to inspect which transcripts you could potentially fail to measure using this protocol. If there are tags for all transcripts, this list will not be produced.
In figure 26.42 you see an example of a table of virtual tags that have been produced using the 3' external option described above.
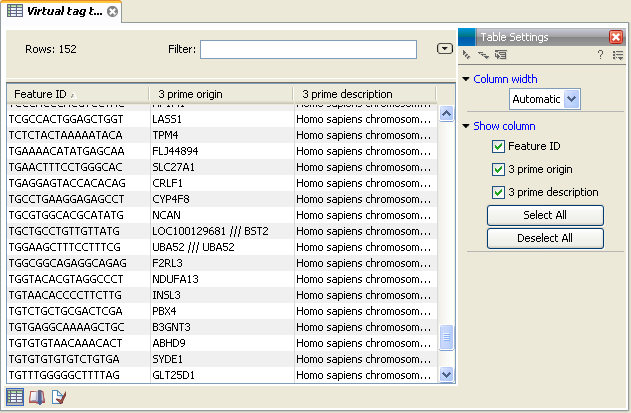
Figure 26.42: A virtual tag table of 3' external tags.
The first column lists the tag itself. This is the column used when you annotate your tag count samples or experiments (see Annotate tag samples or experiment). Next follows the name of the tag's origin transcript. Sometimes the same tag is seen in more than one transcript. In that case, the different origins are separated by /// as it is the case for the tag of LOC100129681 /// BST2 in figure 26.42. The row just below, UBA52, has the same name listed twice. This is because the analysis was based on mRNA annotations from a Refseq genome where each splice variant has its own mRNA annotation, and in this case the UBA52 gene has two mRNA annotations including the same tag.
The last column is the description of the transcript (which is either the sequence description if you use a list of un-annotated sequences or all the information in the annotation if you use annotated sequences).
The example shown in figure 26.42 is the simplest case where only the 3' external tags are listed. If you choose to list All tags, the table will look like figure 26.43.
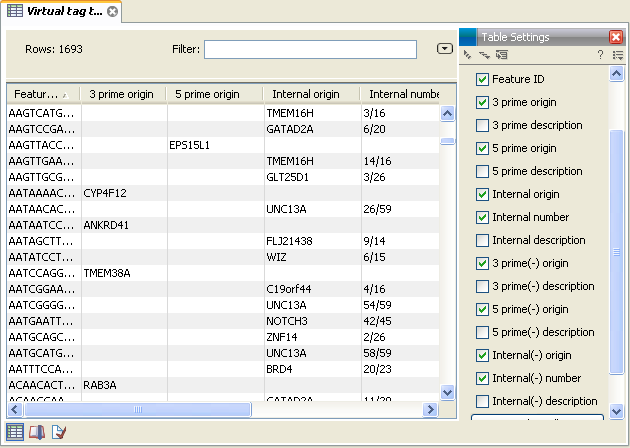
Figure 26.43: A virtual tag table where all tags have been extracted. Note that some of the columns have been ticked off in the Side Panel.
In addition to the information about the 3' tags, there are additional columns for 5' and internal tags. For the internal tags there is also a numbering, see for example the top row in figure 26.43 where the TMEM16H tag is tag number 3 out of 16. This information can be used to judge how close to the 3' end of the transcript the tag is. As mentioned above, you would often expect to sequence more tags from cut sites near the 3' end of the transcript.
If you have chosen to include reverse complemented sequences in the analysis, there will be an additional set of columns for the tags of the other strand, denoted with a (-).
You can use the advanced table filtering (see Working with tables) to interrogate the number of tags with specific origins (e.g. define a filter where 3' origin != and then leave the text field blank).