Expression profiling by tags
Expression profiling by tags, also known as tag profiling or tag-based transcriptomics, is an extension of Serial analysis of gene expression (SAGE) using next-generation sequencing technologies. With respect to sequencing technology it is similar to RNA-seq (see RNA-Seq), but with tag profiling, you do not sequence the mRNA in full length. Instead, small tags are extracted from each transcript, and these tags are then sequenced and counted as a measure of the abundance of each transcript. In order to tell which gene's expression a given tag is measuring, the tags are often compared to a virtual tag library. This consists of the 'virtual' tags that would have been extracted from an annotated genome or a set of ESTs, had the same protocol been applied to these. For a good introduction to tag profiling including comparisons with different micro array platforms, we refer to [t Hoen et al., 2008]. For more in-depth information, we refer to [Nielsen, 2007].
Figure 26.34 shows an example of the basic principle behind tag profiling. There are variations of this concept and additional details, but this figure captures the essence of tag profiling, namely the extraction of a tag from the mRNA based on restriction cut sites.
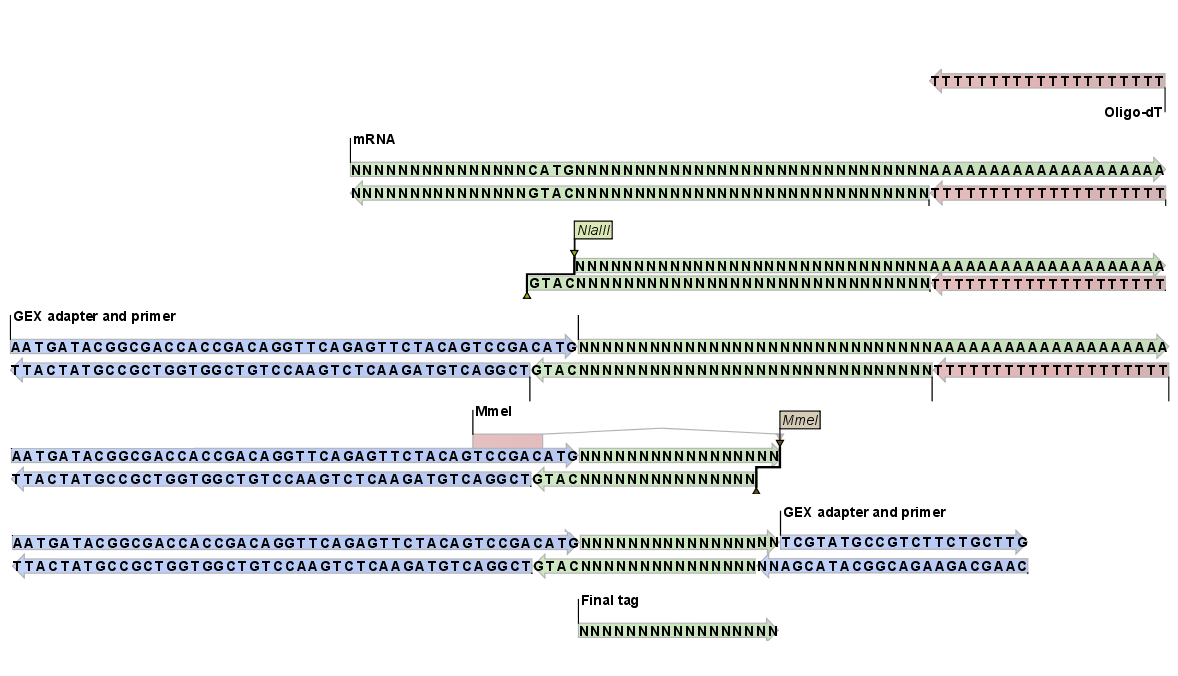
Figure 26.34: An example of the tag extraction process. 1+2. Oligo-dT attached to a magnetic bead is used to trap mRNA. 3. The enzyme NlaIII cuts at CATG sites and the fragments not attached to the magnetic bead are removed. 4. An adapter is ligated to the GTAC overang. 5. The adapter includes a recognition site for MmeI which cuts 17 bases downstream. 6. Another adapter is added and the sequence is now ready for amplification and sequencing. 7. The final tag is 17 bp. The example is inspired by [t Hoen et al., 2008].
The CLC Genomics Workbench supports the entire tag profiling data analysis work flow following the sequencing:
- Extraction of tags from the raw sequencing reads (tags from different samples are often barcoded and sequenced in one pool).
- Counting tags including a sequencing-error correction algorithm.
- Creating a virtual tag list based on an annotated reference genome or an EST-library.
- Annotating the tag counts with gene names from the virtual tag list.
Each of the steps in the work flow are described in details below.
Subsections