Ranking structures
The protein sequence of the gene affected by the variant (the query sequence) is BLASTed against the protein structure sequence database (Download Find Structure Database).
A template quality score is calculated for the available structures found for the query sequence. The purpose of the score is to rank structures considering both their quality and their homology to the query sequence.
The five descriptors contributing to the score are:
- E-value
- % Match identity
- % Coverage
- Resolution (of crystal structure)
- Free R-value (Rfree of crystal structure)
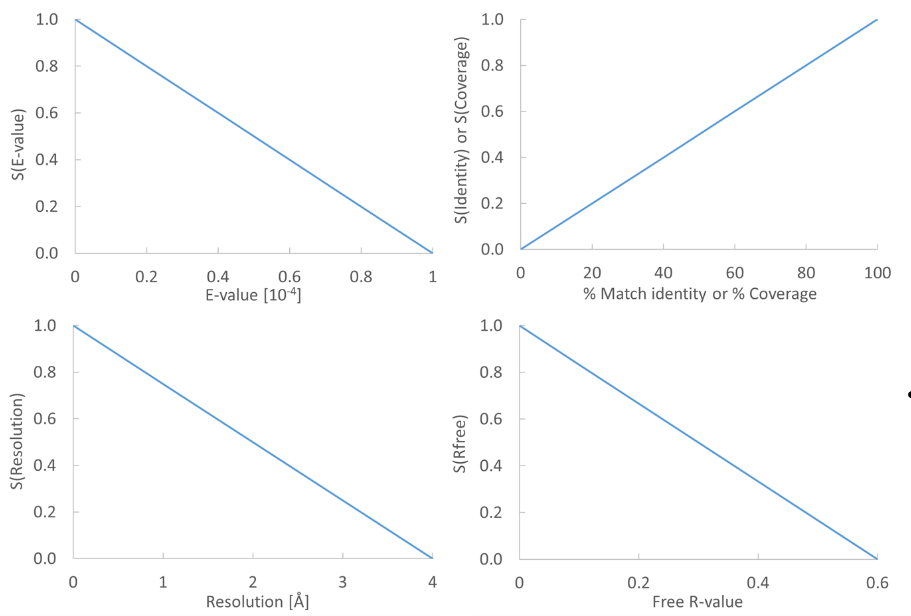
Figure 17.17: From the E-value, % Match identity, % Coverage, Resolution, and Free R-value, the contributions to the "Template quality score" are determined from the linear functions shown in the graphs.
Each of the five descriptors are scaled to [0,1], based on the linear functions seen in figure 17.17. The five scaled descriptors are combined into the template quality score, weighting them to emphasize homology over structure qualities.
E-value is a measure of the quality of the match returned from the BLAST search. You can read more about BLAST and E-values in Bioinformatics explained: BLAST.
% Match identity is the identity between the query sequence and the BLAST hit in the matched region. It is evaluated as
where LB is the length of the BLAST alignment of the matched region, as indicated in figure 17.18, and "Identity in BLAST alignment" is the number of identical positions in the matched region.
% Coverage indicates how much of the query sequence has been covered by a given BLAST hit (see figure 17.18). It is evaluated as
where LG is the total length of gaps in the BLAST alignment and LQ is the length of the query sequence.
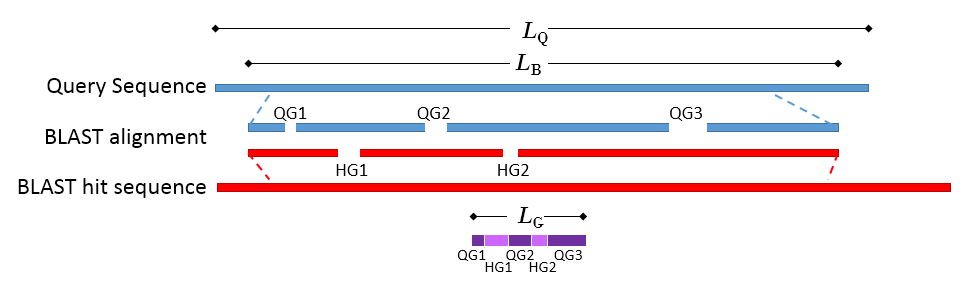
Figure 17.18: Schematic of a query sequence matched to a BLAST hit. LQ is the length of the query sequence, LB is the length of the BLAST alignment of the matched region, QG1-3 are gaps in the matched region of the query sequence, HG1-2 are gaps in the matched region of the BLAST hit sequence, LG is the total length of gaps in the BLAST alignment.
The resolution of a crystal structure is related to the size of structural features that can be resolved from the raw experimental data.
Rfree is used to assess possible overmodeling of the experimental data.
Resolution and Rfree are only given for crystal structures. NMR structures will therefore usually be ranked lower than crystal structures. Likewise, structures where Rfree has not been given will tend to receive a lower rank. This often coincides with structures of older date.