Illumina
CLC Genomics Workbench supports data from Illumina's Genome Analyzer, HiSeq 2000, NextSeq and the MiSeq systems. Choosing the Illumina import will open the dialog shown in figure 6.10. This data type can also be imported using the on-the-fly import functionality described in Launching workflows individually and in batches.
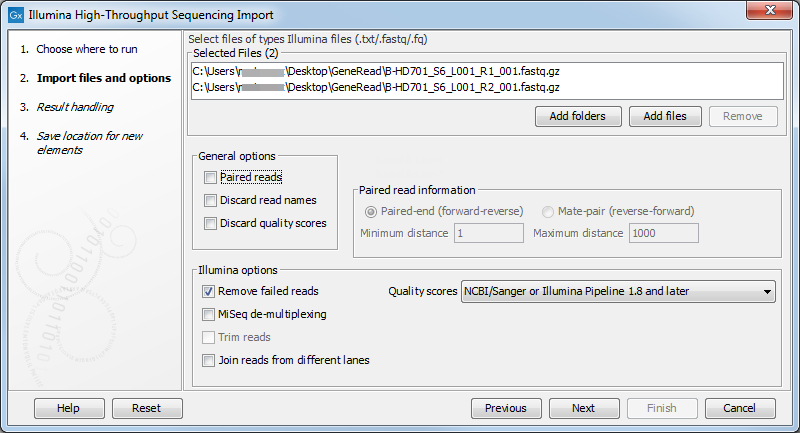
Figure 6.10: Importing data from Illumina systems.
File format
The file formats accepted are Fastq and compressed data in gzip format (*.gz). Paired data in either of these formats can be imported.
Note that there is information inside fastq files specifying whether a read has passed a quality filter or not. If you check Remove failed reads these reads will be ignored during import.
For fastq files, part of the header information for the quality score has a flag where Y means failed and N means passed. In this example, the read has not passed the quality filter:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
Note! In the Illumina pipeline 1.5-1.7, the letter B in the quality score has a special meaning. 'B' is used as a trim clipping. This means that when selecting Illumina pipeline 1.5-1.7, the reads are automatically trimmed when a B is encountered at either end of the reads in the input file. This will happen also if you choose to discard quality scores during import.
General Options
The General options to the left are:
- Paired reads. For paired import, you can select whether the data is Paired-end or Mate-pair. For paired data, the Workbench expects the first reads of the pairs to be in one file and the second reads of the pairs to be in another. So, for example, if you had specified that the pairs were in forward-reverse orientation, then the first file would be assumed to contain the forward reads. The second file would be assumed to contain the reverse reads.
When loading files containing paired data, the CLC Genomics Workbench sorts the files selected according to rules based on the file naming scheme:
- For files coming off the CASAVA1.8 pipeline, we organize pairs according to their identifier and chunk number. Files named with
_R1_are assumed to contain the first sequences of the pairs, and those with_R2_in the name are assumed to contain the second sequence of the pairs. - For other files, we sort them all alphanumerically, and then group them two by two. This means that files 1 and 2 in the list are loaded as pairs, files 3 and 4 in the list are seen as pairs, and so on.
In the simplest case, the files are typically named as shown in figure 6.10. In this case, the data is paired end, and the file containing the forward reads is called
s_1_1_sequence.txtand the file containing reverse reads is calleds_1_2_sequence.txt. Other common filenames for paired data, like_1_sequence.txt,_1_fastq.txt,_2_sequence.txtor_2_fastq.txtwill be sorted alphanumerically. In such cases, files containing the final_1should contain the first reads of a pair, and those containing the final_2should contain the second reads of a pair.For files from CASAVA1.8, files with base names like these: ID_R1_001, ID_R1_002, ID_R2_001, ID_R2_002 would be sorted in this order:
- ID_R1_001
- ID_R2_001
- ID_R1_002
- ID_R2_002
The data in files ID_R1_001 and ID_R2_001 would be loaded as a pair, and ID_R1_002, ID_R2_002 would be loaded as a pair.
Within each file, the first read of a pair will have a
1somewhere in the information line. In most cases, this will be a/1at the end of the read name. In some cases though (e.g. CASAVA1.8), there will be a1elsewhere in the information line for each sequence. Similarly, the second read of a pair will have a2somewhere in the information line - either a/2at the end of the read name, or a2elsewhere in the information line.If you do not choose to discard your read names on import (see next parameter setting), you can quickly check that your paired data has imported in the pairs you expect by looking at the first few sequence names in your imported paired data object. The first two sequences should have the same name, except for a
1or a2somewhere in the read name line.Paired-end and mate-pair data are handled the same way with regards to sorting on filenames. Their data structure is the same the same once imported into the Workbench. The only difference is that the expected orientation of the reads: reverse-forward in the case of mate pairs, and forward-reverse in the case of paired end data. Read more about handling paired data.
- For files coming off the CASAVA1.8 pipeline, we organize pairs according to their identifier and chunk number. Files named with
- Discard read names. For high-throughput sequencing data, the naming of the individual reads is often irrelevant given the huge amount of reads. This option allows you to discard read names to save disk space.
- Discard quality scores. Quality scores are visualized in the mapping view and they are used for SNP detection. If this is not relevant for your work, you can choose to Discard quality scores. One of the benefits from discarding quality scores is that you will gain a lot in terms of reduced disk space usage and memory consumption. Read more about the quality scores of Illumina below.
Paired read information
These options become available if you selected the option "Paired reads" in the General options. First, it is very important to select the correct nature of the paired reads imported: Paired-end (forward-reverse) or Mate-pair (reverse-forward). Second, you have to specify the Minimum and Maximum distances for your pairs. The paired read distance includes the full read sequence, which means that is from the beginning of the forward read to the beginning of the reverse read (figure 6.11). The distances are usually defined during the library preparation of your sequencing experiment, but in doubt you can enter default values: for paired-end the distances are between 1 and 1000 bp while mate-pair reads typically have longer distances between 1000-5000 bp (and sometimes up to 10000). Note that the tools usually used subsequently to process Illumina reads (such as Map Reads to Reference or RNA-Seq Analysis) have an "Auto-detect paired distances" option that is enabled by default. As long as this option is used, mis-specifying the distances during import should bear no consequences.
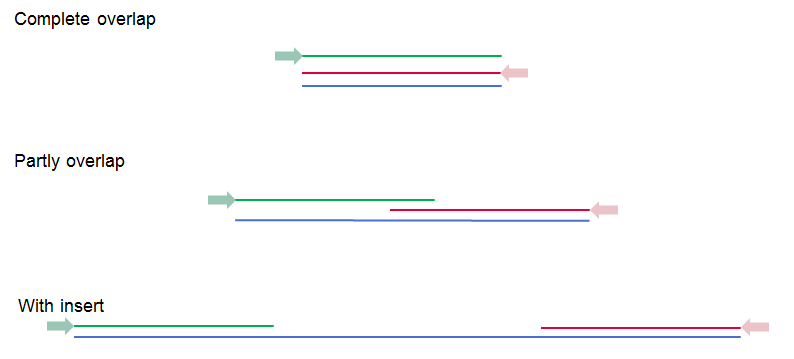
Figure 6.11: Green lines represent forward reads, red lines reverse reads, and in blue is shown the distance of the sequenced DNA fragment. Thus, if there is a complete overlap, the minimum distance will not be 0, but the length of the overlap.
Illumina options
- Remove failed reads. If you check Remove failed reads, reads that did not pass a quality filter (in fastq files) will be ignored during import. For more information on format specific quality filters see section on file format above. If you import paired data and one read in a pair is removed during import, the remaining mate will be saved in a separate sequence list with single reads.
- MiSeq de-multiplexing. Using this option on MiSeq multiplexed data will divide reads into different files based on the "IndexSequence" of the read header:
@Instrument:RunID:FlowCellID:Lane:Tile:X:Y:UMI ReadNum:FilterFlag:0:IndexSequence
Subsequent analysis can then be executed in batch on all the files, and results can be compared at the end.
- Trim reads. This option applies to Illumina Pipeline 1.5 to 1.7. In this pipeline, the value 2 (B) has special meaning and is used as a trim clipping. This means that when selecting Illumina Pipeline 1.5 and later, the reads are trimmed when a B is encountered at either end of the reads in the input file if the Trim reads option is checked.
- Join reads from different lanes. Fastq files from the same sequencing run but from different lanes will be merged into a single sequences list.
Click Next to adjust how to handle the results. We recommend choosing Save in order to save the results directly to a folder, since you probably want to save anyway before proceeding with your analysis. There is an option to put the import data into a separate folder. This can be handy for better organizing subsequent analysis results and for batch processing.
Subsections