QC for Read Mapping
Note that this tool can be used to create a detailed report on a read mapping or a de novo assembly.
To create a detailed mapping report:
Toolbox | Quality Control (![]() ) | QC for Read Mapping (
) | QC for Read Mapping (![]() )
)
This opens a dialog where you can select mapping results (![]() )/ (
)/ (![]() )/ (
)/ (![]() ), simples contigs from a de novo assembly, or RNA-Seq analysis results (
), simples contigs from a de novo assembly, or RNA-Seq analysis results (![]() ).
).
In the next wizard window "Set contig group" (figure 26.11), you can set thresholds for grouping long and short contigs. Inputs for which thresholds can be specified are simple contigs or stand-alone mappings generated by a tool that works with de novo assemblies. These options are disabled and greyed out if you are working with a read mapping or an RNA-Seq analysis mapping.
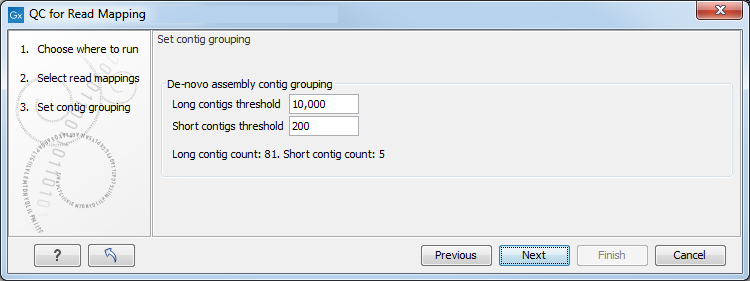
Figure 26.11: Parameters for mapping reports.
The grouping is used to show statistics (e.g., number of contigs, mean length) for the contigs in each group. Note that the de novo assembly in the CLC Genomics Workbench per default only reports contigs longer than 200 bp (this can be changed when running the assembly).
In the last dialog (figure 26.12), by checking "Create table with statistics for each mapping", you can create a table showing detailed statistics for each reference sequence (for de novo results the contigs act as reference sequences, so it will be one row per contig).
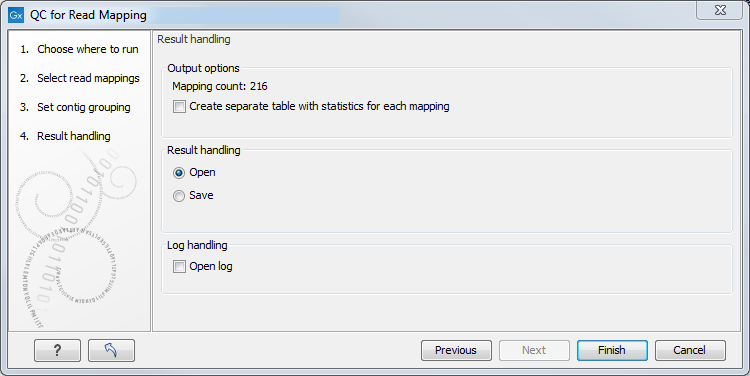
Figure 26.12: Result handling options.
The first section of the detailed mapping report is a summary of the statistics:
- Reference count
- Type
- Total reference length
- GC contents in %
- Total read count
- Mean read length
- Total read length
The rest of the report, as well as the optional statistic tables are described in the following sections.
Subsections