Demultiplex Reads
Multiplexing techniques are often used when sequencing different samples in one sequencing run. One method used is to tag the sequences with a unique identifier during the preparation of the sample for sequencing [Meyer et al., 2007].
With this technique, each sequence read will have a sample-specific tag, which is a specific sequence of nucleotides before and after the sequence of interest. This principle is shown in figure 25.16.
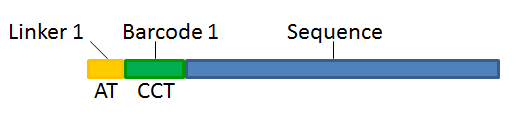
Figure 25.16: Tagging the target sequence, which in this case is single reads from one sample.
The sample-specific tag, also called the barcode or the index, can then be used to distinguish between the different samples when analyzing the sequencing data.
Post-processing of the sequencing data is required to separate the reads into their corresponding samples. Based on their barcodes this can be done using the demultiplexing functionality of the CLC Genomics Workbench. Using this tool, sequences are associated with a particular sample when they contain an exact match to a particular barcode. Sequences that do not contain an exact match to any of the barcode sequences provided are classified as not grouped and are put into a sequence list with the name "Not grouped".
Note that there is also an example using Illumina data in here.
Before processing the data, you need to import it as described in Import high-throughput sequencing data.
Please note that demultiplexing is often carried out on the sequencing machine so that the sequencing reads are already separated according to sample. This is often the best option, if it is available to you. Of course, in such cases, the data will not need to be demuliplexed again after import into the CLC Genomics Workbench.
Subsections
- Demultiplexing single reads
- Demultiplexing paired reads
- Entering barcodes
- Demultiplexing output options
- An example using Illumina barcoded sequences