Ploidy state detection
Detect Regional Ploidy is inspired by [Beroukhim et al., 2006].
The tool predicts a ploidy state (table 10.1) for each locus from the input tracks: a CNV target or a somatic SNP that
- Is assumed to be heterozygous in normal cells.
- Overlaps a target region from the CNV track.
A ploidy state can be associated to loss-of-heterozygosity (LOH), which is characterized by loss of one allele, whereas the other allele is present in one or more copies.
The tool uses as evidence for copy-number changes the relative log coverage ratio (RLR), calculated as the signed ![]() of the absolute adjusted fold-change from the CNV track.
of the absolute adjusted fold-change from the CNV track.
The tool also relies on B-allele frequencies for somatic SNPs that are assumed to be heterozygous in normal cells. For this, either matched germline variants or a variant database are needed:
- Germline variants. Germline variants detected from a matched normal sample. The provided variant track is automatically filtered to heterozygous variants.
- Variant database. When a matched normal samples is not available, a variant database can be used.
If either somatic or germline variants have the "Filter" attribute set (see Filter on Custom Criteria), only variants where this attribute is set to PASS are used.
The sample purity, the proportion of cells in the sample that are tumor-derived, and the ploidy state of a locus determine the expected RLR and B-allele frequencies of the heterozygous variants. For example, consider a sample with 60% purity, where the tumor cells contain a duplication in a locus with two alleles, A and B.
- The expected B-allele frequency is calculated as the support for the B-allele relative to the total allelic support. If 100 cells are sequenced uniformly:
- The 60 tumor cells contains two copies of A and one copy of B, and
- The 40 normal cells contains one copy of A and one copy of B,
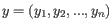 .
.
- The expected RLR is calculated by comparing the read coverage to a normal diploid sample. If the normal sample yields 200 reads, the 60% tumor sample would yield 260 reads:
- The 60% tumor cells yield
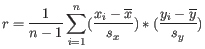 reads due to the duplication.
reads due to the duplication.
- The 40% normal cells yield
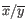 reads.
reads.
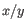 and an RLR of
and an RLR of
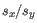 .
.
- The 60% tumor cells yield
The tool jointly optimizes:
- The sample purity.
- A normalization factor if Normalize coverage is checked.
- A Hidden Markov Model (HMM) containing the nine ploidy states as the hidden states.
The normalization factor helps correct for systematic coverage shifts. For example, if a large fraction of loci are affected by a deletion, the RLR may be too low, resulting in under-detection of copy-number changes. Because deletions affect both coverage and allele frequencies, the model uses the observed B-allele frequencies to adjust the RLR appropriately. For example, in a case where a normal sample has copy number 2 and a tumor sample with copy number 1 throughout, the normalization factor should ideally be 0.5.
The optimized parameters and HMM are then used to filter loci and generate regions by the following steps:
- The HMM is used to predict the most likely ploidy state for each locus. Neighboring loci with the same ploidy state are then merged into contiguous regions.
- If Remove outliers is checked:
- Loci whose RLR or B-allele frequency is more than three standard deviations from the mean of their region are excluded. SNPs are only removed from regions with at least 10 SNPs, and CNVs are only removed from regions with at least 10 CNV targets to avoid unstable calculations of standard deviations.
- Subsequently, the parameter optimization, ploidy state prediction, and merging of loci into regions is repeated for the remaining loci.
- Regions originating from fewer loci than Minimum loci count are removed. Merging is then repeated, in case this removal leads to new neighboring regions with the same ploidy state.
- Regions are extended to nearby boundaries when the distance (in megabases) to the boundary is smaller than Maximum region distance (Mb). The following, whichever is closest, is used:
- The chromosome start or end.
- The centromere.
- Neighboring region. In this situation, both regions are extended to the midpoint between them.
This helps reduce small gaps between regions in areas without informative loci.
- Regions shorter than Minimum length (Mb) are removed. Merging is then repeated, in case this removal leads to new neighboring regions with the same state.
Finally, loci and regions are annotated with an LOH status, according to table 10.1 and are output in a locus-level ploidy track and a region-level ploidy track, respectively.
Limitations
Detect Regional Ploidy is designed for autosomal chromosomes. The underlying model does not account for the haploid baseline of sex chromosomes in male samples, and may therefore misinterpret the coverage and allele frequencies in these regions. Results for sex chromosomes should be interpreted with caution.