Gap costs
The alignment algorithm has three parameters
concerning gap costs: Gap open cost, Gap extension cost and End gap
cost. The precision of these parameters is to one place of decimal.
- Gap open cost. The price for introducing gaps in an alignment.
- Gap extension cost. The price for every extension past the initial gap.
However, for most alignments it is a good idea to make the Gap open cost quite a bit higher than the Gap extension cost. The default values are 10.0 and 1.0 for the two parameters, respectively.
- End gap cost. The price of gaps at the beginning or the end of the alignment. One of the advantages of the CLC Genomics Workbench alignment method is that it provides flexibility in the treatment of gaps at the ends of the sequences. There are three possibilities:
Many homologous proteins have quite different ends, often with large insertions or deletions. This confuses alignment algorithms, but using the Cheap end gaps option, large gaps will generally be tolerated at the sequence ends, improving the overall alignment. This is the default setting of the algorithm.
Finally, treating end gaps like any other gaps is the best option when you know that there are no biologically distinct effects at the ends of the sequences.
Figures 20.3 and 20.4 illustrate the differences between the different gap scores at the sequence ends.

Figure 20.3: The first 50 positions of two different alignments of seven calpastatin sequences. The top alignment is made with cheap end gaps, while the bottom alignment is made with end gaps having the same price as any other gaps. In this case it seems that the latter scoring scheme gives the best result.
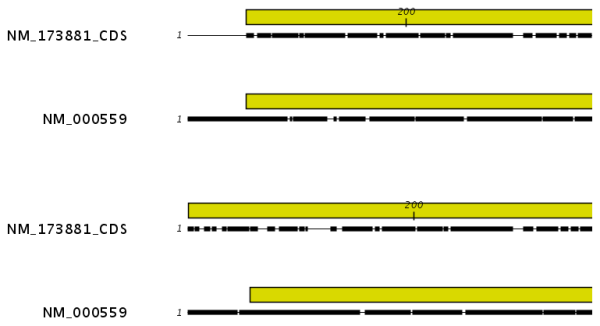
Figure 20.4: The alignment of the coding sequence of bovine myoglobin with the full mRNA of human gamma globin. The top alignment is made with free end gaps, while the bottom alignment is made with end gaps treated as any other. The yellow annotation is the coding sequence in both sequences. It is evident that free end gaps are ideal in this situation as the start codons are aligned correctly in the top alignment. Treating end gaps as any other gaps in the case of aligning distant homologs where one sequence is partial leads to a spreading out of the short sequence as in the bottom alignment.