Amino acid changes
This tool annotates variants with amino acid changes and creates a track for visual inspection of the amino acid changes. It takes a variant track as input and also requires a track with coding regions and a reference sequence.
To add information about amino acid changes to a variant track:
Toolbox | Resequencing Analysis (![]() ) | Functional Consequences (
) | Functional Consequences (![]() ) | Amino Acid Changes (
) | Amino Acid Changes (![]() )
)
If you are connected to a server, the first wizard step will ask you where you would like to run the analysis. Next, you must provide the variant track to be annotated with amino acid changes (see figure 27.64).
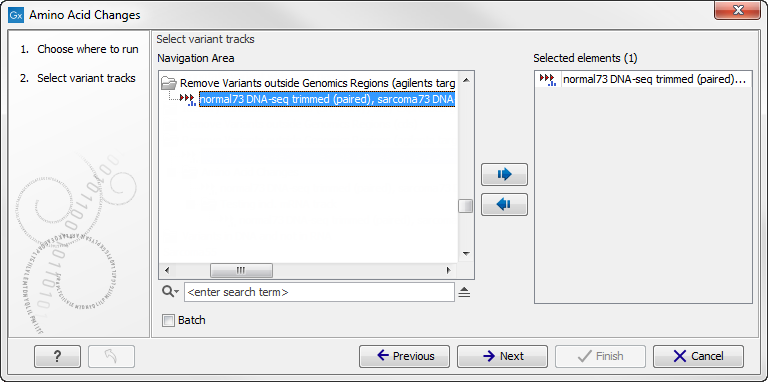
Figure 27.64: The amino acid changes annotation tool takes variant tracks as input.
Click on the button labeled Next to go to the next wizard step (figure 27.65).
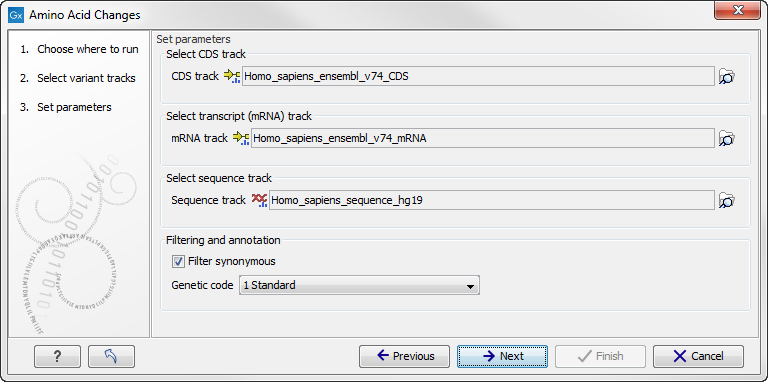
Figure 27.65: Select CDS, mRNA, and sequence track and choose whether or not you would like to filter away synonymous variants.
In this step you get the following options:
- Select CDS track Click on the folder icon (
 ) in the right side of the wizard to select a CDS track. The CDS track is used to determine the reading frame and exon location to be used for translation. If you do not already have CDS, mRNA, and sequence tracks in the Workbench, you can download it by clicking on the Download icon (
) in the right side of the wizard to select a CDS track. The CDS track is used to determine the reading frame and exon location to be used for translation. If you do not already have CDS, mRNA, and sequence tracks in the Workbench, you can download it by clicking on the Download icon ( ) found in the upper right corner of the Workbench. Select Download Reference Genome Data (
) found in the upper right corner of the Workbench. Select Download Reference Genome Data ( ) as described in Download reference genome data.
) as described in Download reference genome data.
- Select mRNA track Click on the folder icon () in the right side of the wizard to select the mRNA track. The mRNA track is used to determine whether the variant is inside or outside the region covered by the transcript. The mRNA track is optional. If you choose to provide an mRNA track you annotate variants that are located in the mRNA but also outside the region covering the coding sequence, in cases where such variants have been detected. If you choose not to provide an mRNA track, variants found outside the CDS region will not be annotated.
- Select sequence track Click on the folder icon () in the right side of the wizard to select the reference sequence track.
- Filtering and annotation The "Filter synonymous" option allows you to filter away synonymous variants that does not cause any change to the encoded amino acid. The genetic code is the code that is used for amino acid translation (see http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi). The default option is "1 standard", the vertebrate standard code. If relevant, you can use the drop-down list to change to the genetic code that applies to you organism.
- Filter CDS regions with no variants When choosing this option, "empty" CDS regions (CDS regions with no overlapping variants) will be filtered away from the track.
Click on the button labeled Next, choose whether you would like to open or save the results and click on the button labeled Finish.
Two types of outputs are generated:
- A variant track that has been annotated with the amino acid changes. The added information can be accessed via the tooltips in the variant track or in the extra columns that have been added to the variant table. The extra columns provide information about the amino acid changes (see http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi). The variant track opens in track view and the table view can be accessed by clicking on the table icon found in the lower left corner of the View Area.
- An amino acid track that displays a graphical presentation of the amino acid changes. The track is based on the CDS track and in addition to the amino acid sequence of the coding sequence, all amino acids that have been affected by variants are shown as individual amino acids below the amino acid track. Changes causing a frameshift are symbolized with two arrow heads, and variants causing premature stop are marked with an asterisk. An example is shown in figure 27.66.
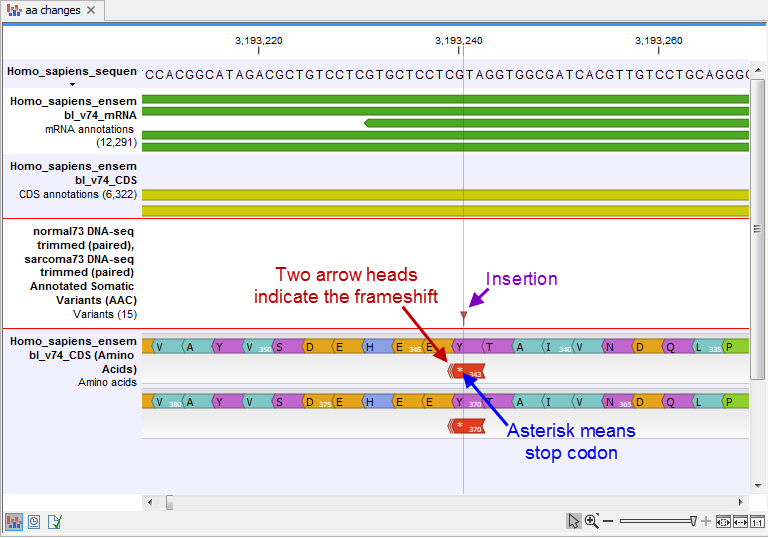
Figure 27.66: The variant track and the amino acid track is here presented together with the reference sequence and the mRNA and CDS tracks. An insertion (purple arrow) has caused a frameshift (red arrow) that has changed an alanine to a stop codon (blue arrow).
For each variant in the input track, the following information is added:
- Coding region change. This describes the relative position on the coding DNA level, using the nomenclature proposed at http://www.hgvs.org/mutnomen/. Variants inside exons and in the untranslated regions of the transcript will also be annotated with the distance to the nearest exon. E.g. "c.-4A>C" describes a SNV four bases upstream of the start codon, while "c.*4A>C" describes a SNV four bases downstream of the stop codon.
- Amino acid change. This describes the change on the protein level. For example, single amino-acid changes caused by SNVs are listed as "p.[Gly261Cys]", denoting that in the protein sequence (hence the "p.") the Glycine at position 261 is changed into Cysteine. Frame-shifts caused by nucleotide insertions and deletions are listed with the extension fs, for example p.[Pro244fs] denoting a frameshift at position 244 coding for Proline. For further details of the nomenclature see the "Recommendations for the description of protein sequence variants (v2.0)" at http://www.hgvs.org/mutnomen/.
- Coding region change in longest transcript. When there are many transcript variants for a gene, the coding region change for all transcripts are listed in the "Coding region change" column. For quick reference, the longest transcript is often used, and there is a special column only listing the coding region change for the longest transcript.
- Amino acid change in longest transcript. This is similar to the above, just on the protein level.
- Other variants within codon. If there are other variants within the same codon, this column will have a "Yes". In this case, it should be manually investigated whether the two variants are linked by reads and the amino acid change annotated by the amino acid changes may not be correct in this case.
- Non-synonymous. Will have a "Yes" if the variant is non-synonymous at the protein level for any transcript. If the filter "Filter synonymous" was applied, this column will only contain entries labeled "Yes".
An example of the output is given in figure 27.67.
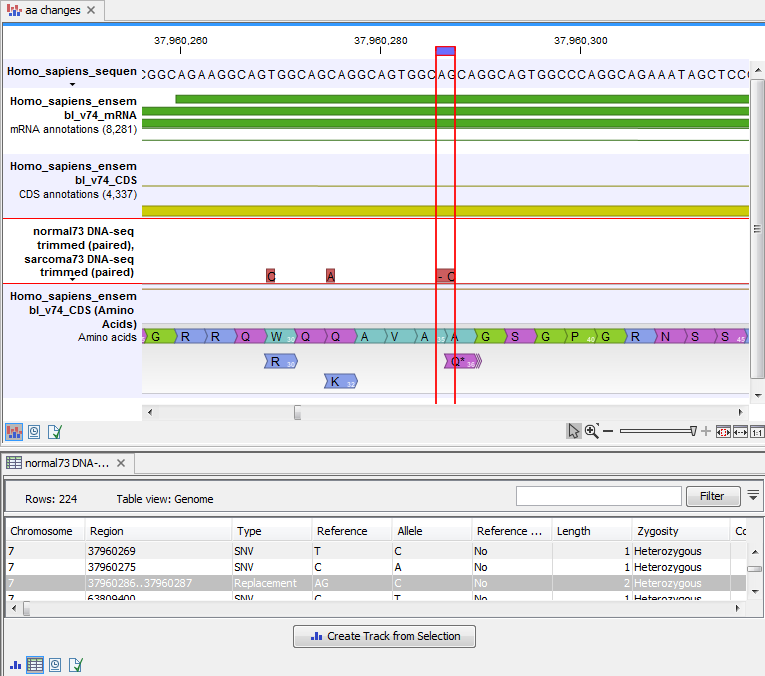
Figure 27.67: The resulting amino acid changes in track and table views. When the variant table has been opened by double-clicking on the text found in the left side of the View Area, the variant table and the variant track are linked. When clicking on an entry in the table, this position will be brought into focus in the variant track.
The top track view displays a track list with the reference sequence, mRNA, CDS, variant, and amino acid tracks. The lower table view is the variant table that has been opened from the track list by double-clicking on the variant track name found in the left-hand side of the View Area. When opening the variant table in split view from the track list, the table and the variant track are linked.
An example illustrating a situation where different variants affect the same codon is shown in figure 27.68.
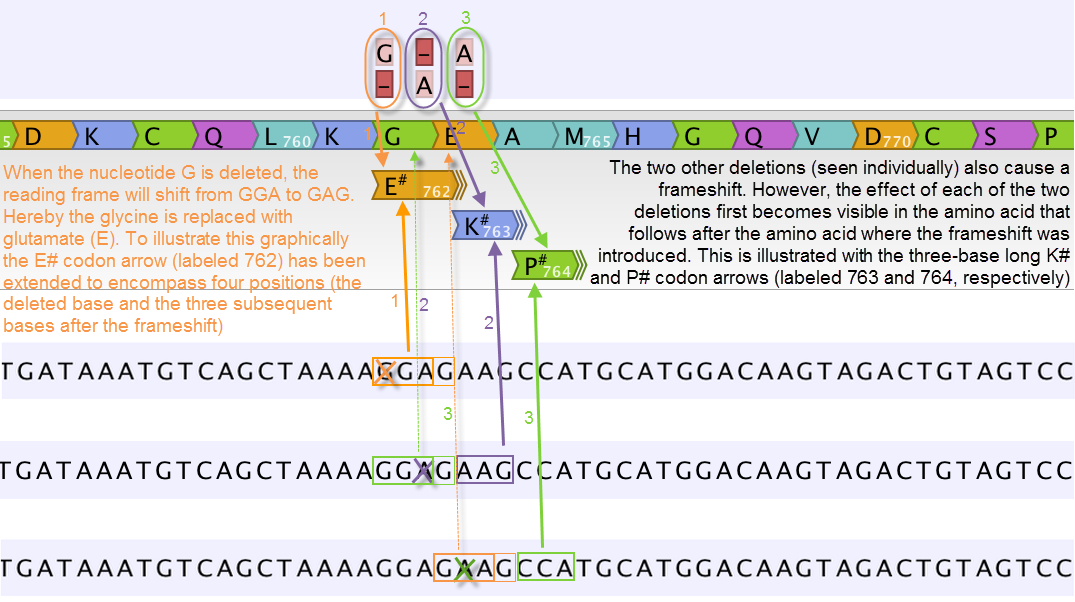
Figure 27.68: Amino acids encoded from codons that potentially could have been affected by more than one variant are marked with a hash symbol (#) as the graphically presented amino acid changes always only include a single variant (a SNV, MNV, insertion, or deletion). Shown here are three different variants, present only one at the time, and the consequences of the three individual deletions. In cases where the deletion is found in a codon that is affected with an amino acid change, the arrow also include the deletion (situation 1) in the two other scenarios, the codon containing the deletion is changed to a codon that encodes the same amino acid, and the effect is therefore not seen until in the subsequent amino acid.
In this example three single nucleotide deletions are shown along with the resulting amino acid changes based on scenarios where only one deletion is present at the time. The first affected amino acid is shown for each of the three deletions. As the first deletion affect the encoded amino acid, this amino acid change is shown with a four nucleotide long arrow (that includes the deletion). The other two deletions do not affect the encoded amino acid as the frameshift was "synonymous" at the position encoded by the codon where the deletion was introduced. The effect is first seen at the next amino acid position (763 and 764, respectively), which does not contain a deletion, and therefore is illustrated with a three nucleotide long arrow.
The hash symbol (#) on the changed amino acids symbolize that more than one variant can be present in the region encoding this specific amino acid. The simultaneous presence of multiple variants within the same codon is not predicted by the amino acid changes tool. Manual inspection of the reads is required to be able to detect multiple variants within one codon.
The amino acid track
The colors of the amino acids in the amino acid track can be changed in the Side Panel under Track layout and "Amino acids track" (see figure 27.69).
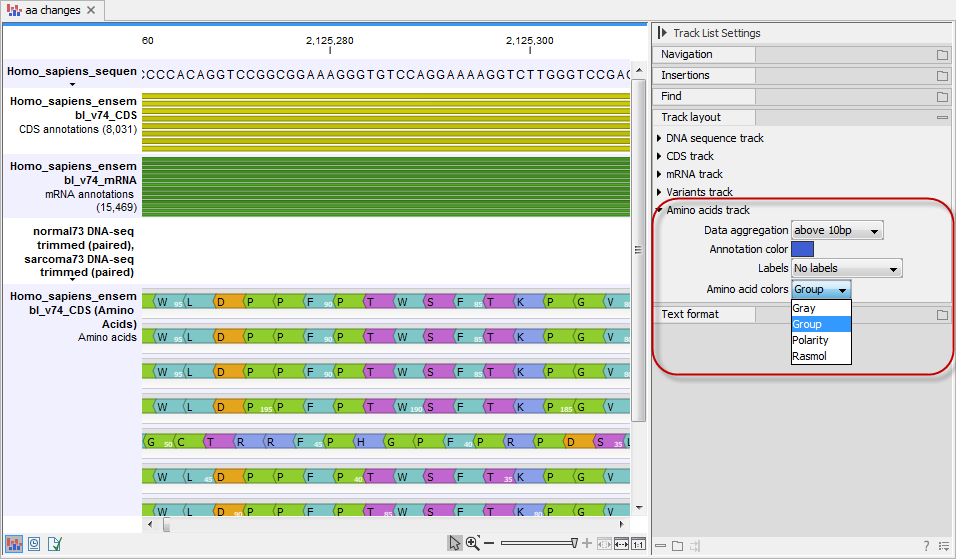
Figure 27.69: The colors of the amino acids can be changed in the Side Panel under "Amino acids track".
Four different color schemes are available under "Amino acid colors":
- Gray All amino acids are shown in gray.
- Group Colors the amino acids in groups by the following properties:
- Purple neutral, polar
- Turquoise neutral, nonpolar
- Orange acidic, polar
- Blue basic ,polar
- Bright green other (functional properties)
- Polarity Colors the amino acids according to the following categories:
- Green neutral, polar
- Black neutral, nonpolar
- Red acidic, polar
- Blue basic ,polar
- Rasmol Colors the amino acids according to the Rasmol color scheme (see http://www.openrasmol.org/doc/rasmol.html).