Merge overlapping pairs
Some paired end library preparation methods using relatively short fragment size will generate data with overlapping pairs.
This type of data can be handled as standard paired-end data in the CLC Genomics Workbench,
and it will work perfectly fine (see details for variant detection in Detailed information about overlapping paired reads).
However, in some situations it can be useful to merge the overlapping pair into one sequence read instead. The benefit is that you get longer reads, and that the quality improves (normally the quality drops towards the end of a read, and by overlapping the ends of two reads, the consensus read now reflects two read ends instead of just one).
In the CLC Genomics Workbench, there is a tool for merging overlapping reads, which are in forward-reverse orientation:
Toolbox | NGS Core Tools (![]() ) | Merge Overlapping Pairs (
) | Merge Overlapping Pairs (![]() )
)
Select one or more sequence lists with paired end sequencing reads as input.
Please note that read pairs have to be in forward-reverse orientation. Please also note that after merging the merged reads will always be in the forward orientation. As an aside, length trimming of reads can be done before or after merging, however the merged read's 3' is the 5' of the reverse read, so that trimming the original reads using the Remove 5' terminal nucleotides option corresponds to trimming the merged reads using both the Remove 5' terminal nucleotides option and the Remove 3' terminal nucleotides option.
Clicking Next allows you to set parameters as displayed in figure 23.32.
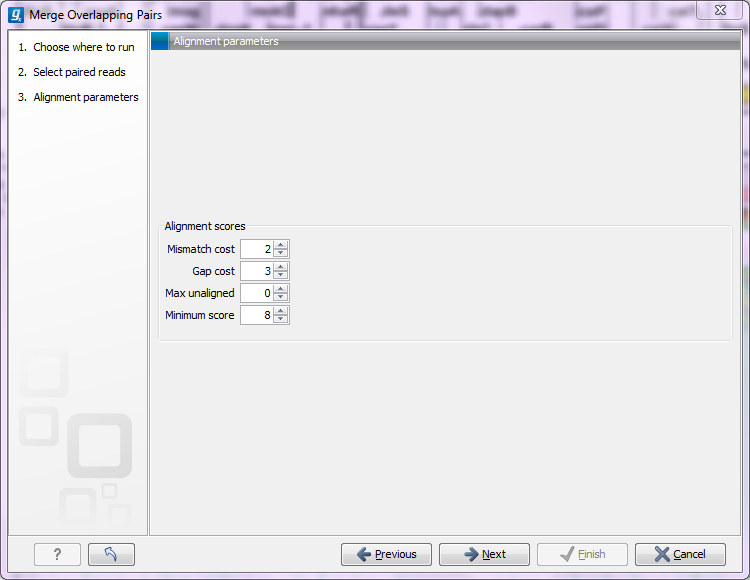
Figure 23.32: Setting parameters for merging overlapping pairs.
In order to understand how these parameters should be set, an explanation of the merging algorithm is needed: Because the fragment size is not an exact number of base pairs and is different from fragment to fragment, an alignment of the two reads has to be performed. If the alignment is good and long enough, the reads will be merged. Good enough in this context means that the alignment has to satisfy some user-specified score criteria (details below). Because of sequencing errors that typically are more abundant towards the end of the read, the alignment is not expected always to be perfect, and the user can decide how many errors are acceptable. Long enough in this context means that the overlap between the reads has to be non-coincidental. Merging two reads that do not really overlap, leads to errors in the downstream analysis, thus it is very important to make sure that the overlap is big enough. If only a few bases overlap was required, some read pairs will match by chance, so this has to be avoided.
The following parameters are used to define what is good enough and long enough
- Mismatch cost The alignment awards one point for a match, and the mismatch cost is set by this parameter. The default value is 2.
- Gap cost This is the cost for introducing an insertion or deletion in the alignment. The default value is 3.
- Max unaligned end mismatches The alignment is local, which means that a number of bases can be left unaligned. If the quality of the reads is dropping to be very poor towards the end of the read, and the expected overlap is long enough, it makes sense to allow some unaligned bases at the end. However, this should be used with great care which is why the default value is 0. As explained above, a wrong decision to merge the reads leads to errors in the downstream analysis, so it is better to be conservative and accept fewer merged reads in the result.
- Minimum score This is the minimum score of an alignment to be accepted for merging. The default value is 10. As an example: with default settings, this means that an overlap of 13 bases with one mismatch will be accepted (12 matches minus 2 for a mismatch).
Please note that even with the alignment scores above the minimum score specified in the tool setup, the paired reads also need to have the number of end mismatches below the "Maximum unaligned end mismatches" value specified in the tool setup to be qualified for merging.
After clicking Next you can select whether a report should be generated as part of the output. The main result will be two sequence lists for each list in the input: one containing the merged reads (marked as single end reads), and one containing the reads that could not be merged (still marked as paired data). Since the CLC Genomics Workbench handles a mix of paired and unpaired data, both of these sequence lists can be used in the further analysis. However, please note that low quality can be one of the reasons why a pair cannot be merged. Hence, the list of reads that could not be paired is more likely to contain more reads with errors than the one with the merged reads.
Subsections