Optimization of the graph using paired reads
When paired reads are available, we can use the paired information to resolve large repeat regions that are not spanned by individual reads, but are spanned by read pairs. Given a set of paired reads that align to two nodes connected by a repeat region, the repeat region may be resolved for those nodes if we can find a path connecting the nodes with a length corresponding to the paired read distance. However, such a path must be supported by a minimum of four sets of paired reads before the repeat is resolved.
If it's not possible to resolve the repeat, scaffolding is performed where paired read information is used to determine the distances between contigs and the orientation of these. Scaffolding is only considered between two contigs if both are at least 120 bp long, to ensure that enough paired read information is available. An iterative greedy approach is used when performing scaffolding where short gaps are closed first, thus increasing the paired read information available for closing gaps (see figure 29.12).
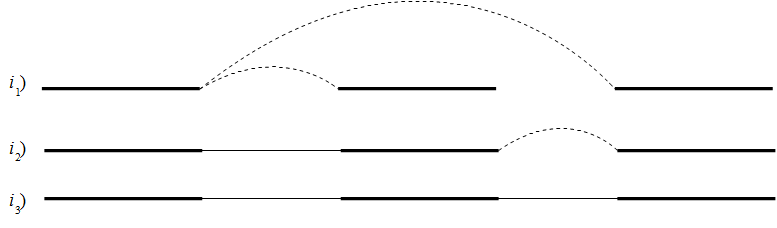
Figure 29.12: Performing iterative scaffolding of the shortest gaps allows long pairs to be optimally used. i1 shows three contigs with dashed arches indicating potential scaffolding. i2 is after first iteration when the shortest gap has been closed and long potential scaffolding has been updated. i3 is the final results with three contigs in one scaffold.
Contigs in the same scaffold are output as one large contig with Ns inserted in between. The number of Ns inserted correspond to the estimated distance between contigs, which is calculated based on the paired read information. More precisely, for each set of paired reads spanning two contigs a distance estimate is calculated based on the supplied distance between the reads. The average of these distances is then used as the final distance estimate. The distance estimate will often be negative which happens when the paired information indicate that two contigs overlap. The assembler will attempt to align the ends of such contigs and if a high quality overlap is found the contigs are joined into a single contig. If no overlap is found, the distance estimate is set to two so that all remaining scaffolds have positive distance estimates.
Furthermore, Ns can also be present in output contigs in cases where input sequencing reads themselves contain Ns.
Please note that in CLC Genomics Workbench 6.0.1, Genomics Server 5.0.1, Assembly Cell 4.0.2 and all earlier versions of these products a performance optimization gave rise to Ns being inserted in certain non-scaffold regions, which in the current version can be solved with reads covering such specific regions.
Additional information about repeats being resolved using paired reads and scaffolded contigs is available as annotations on the contig sequences and as summary in the report (see De novo assembly report). This information can also be exported in AGP format (see AGP export).
The annotations in table format can be viewed by clicking the "Show Annotation Table" icon (![]() ) at the bottom of the viewing area. "Show annotation types" in the side panel allows you to select the annotation "Scaffold" among a list of other annotations. The annotations tell you about the scaffolding that was performed by the de novo assembler. That is, it tells you where particular contigs, those areas containing complete sequence information, were joined together across regions without complete sequence information.
) at the bottom of the viewing area. "Show annotation types" in the side panel allows you to select the annotation "Scaffold" among a list of other annotations. The annotations tell you about the scaffolding that was performed by the de novo assembler. That is, it tells you where particular contigs, those areas containing complete sequence information, were joined together across regions without complete sequence information.
For the GFF format there are three types of annotations:
- Scaffold refers to the estimated gap region between two contigs where Ns are inserted.
- Contigs joined refers to the join of two contigs connected by a repeat or another ambiguous structure in the graph, which was resolved using paired reads. Can also refer to overlapping contigs in a scaffold that were joined using an overlap.
- Alternatives excluded refers to the exclusion of a region in the graph using paired reads, which resulted in a join of two contigs.