What do the SignalP scores mean?
Many bioinformatics approaches or prediction tools do not give a yes/no answer. Often the user is facing an interpretation of the output, which can be either numerical or graphical. Why is that? In clear-cut examples there are no doubt; yes: this is a signal peptide! But, in borderline cases it is often convenient to have more information than just a yes/no answer. Here a graphical output can aid to interpret the correct answer. An example is shown in figure 16.5.
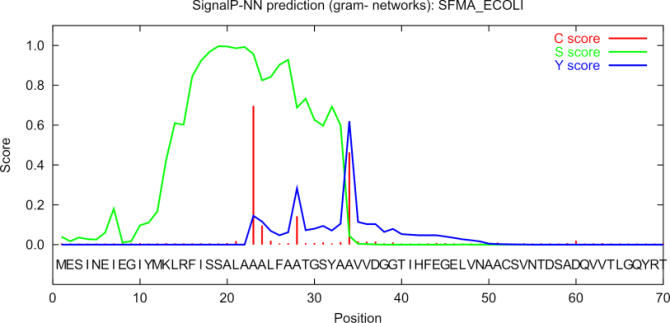
Figure 16.5: Graphical output from the SignalP method of
Swiss-Prot entry SFMA_ECOLI. Initially this seemed like
a borderline prediction, but closer inspection of the sequence
revealed an internal methionine at position 12, which could
indicate a erroneously annotated start of the protein. Later this
protein was re-annotated by Swiss-Prot to start at the M in
position 12. See the text for description of the scores.
The graphical output from SignalP (neural network) comprises three different scores, C, S and Y. Two additional scores are reported in the SignalP3-NN output, namely the S-mean and the D-score, but these are only reported as numerical values.
For each organism class in SignalP; Eukaryote, Gram-negative and Gram-positive, two different neural networks are used, one for predicting the actual signal peptide and one for predicting the position of the signal peptidase I (SPase I) cleavage site. The S-score for the signal peptide prediction is reported for every single amino acid position in the submitted sequence, with high scores indicating that the corresponding amino acid is part of a signal peptide, and low scores indicating that the amino acid is part of a mature protein.
The C-score is the "cleavage site" score. For each position in the submitted sequence, a C-score is reported, which should only be significantly high at the cleavage site. Confusion is often seen with the position numbering of the cleavage site. When a cleavage site position is referred to by a single number, the number indicates the first residue in the mature protein. This means that a reported cleavage site between amino acid 26-27 corresponds to the mature protein starting at (and include) position 27.
Y-max is a derivative of the C-score combined with the S-score resulting in a better cleavage site prediction than the raw C-score alone. This is due to the fact that multiple high-peaking C-scores can be found in one sequence, where only one is the true cleavage site. The cleavage site is assigned from the Y-score where the slope of the S-score is steep and a significant C-score is found.
The S-mean is the average of the S-score, ranging from the N-terminal amino acid to the amino acid assigned with the highest Y-max score, thus the S-mean score is calculated for the length of the predicted signal peptide. The S-mean score was in SignalP version 2.0 used as the criteria for discrimination of secretory and non-secretory proteins.
The D-score is introduced in SignalP version 3.0 and is a simple average of the S-mean and Y-max score. The score shows superior discrimination performance of secretory and non-secretory proteins to that of the S-mean score which was used in SignalP version 1 and 2.
For non-secretory proteins all the scores represented in the SignalP3-NN output should ideally be very low.
The hidden Markov model calculates the probability of whether the submitted sequence contains a signal peptide or not. The eukaryotic HMM model also reports the probability of a signal anchor, previously named uncleaved signal peptides. Furthermore, the cleavage site is assigned by a probability score together with scores for the n-region, h-region, and c-region of the signal peptide, if it is found.
Other useful resources
http://www.cbs.dtu.dk/services/SignalP
Pubmed entries for some of the original papers.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=AbstractPlus&list_uids=9051728&query_hl=1&itool=pubmed_docsum