Mapping in color space
Reads from a SOLiD sequencing run may exhibit all the same differences to a reference sequence as reads from other technologies: mismatches, insertions and deletions. On top if this, SOLiD reads may exhibit color errors, where a color is read wrongly and the rest of the read is affected. If such an error is detected, it can be corrected and the rest of the read can be converted to what it would have been without the error.Consider this SOLiD read:
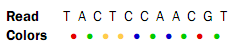
The first nucleotide (T) is from the primer, so this is ignored in the following analysis. Now, assume that a reference sequence is this:
![]()
Here, the colors are just inferred since they are not the result of a sequencing experiment.
Looking at the colors, a possible alignment presents itself:
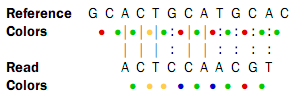
In the beginning of the read, the nucleotides match (ACT), then there is a mismatch (G in reference and C in read), then two more matches (CA), and finally the rest of the read does not match. But, the colors match at the end of the read. So a possible interpretation of the alignment is that there is a nucleotide change in position four of the read and a color space error between positions six and seven in the read. Such an interpretation can be represented as:
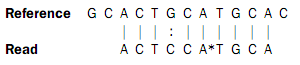
Here, the * represents a color error. The remaining part of the displayed read sequence has been adjusted according to the inferred error. So this alignment scores nine times the match score minus the mismatch cost and a color error cost. This color error cost is a new parameter that is introduced when performing read mapping in color space.
Note that a color error may be inferred before the first nucleotide of a read. This is the very first color after the known primer nucleotide that is wrong, changing the whole read.
Here is an example from a set of real SOLiD data that was reference assembled by taking color space into account using ungapped global alignments.
444_1840_767_F3 has 1 match with a score of 35:
1046535 GATACTCAATGCCGCCAAAGATGGAAGCCGGGCCA 1046569 reference
|||||||||||||||||||||||||||||||||||
GATACTCAATGCCGCCAAAGATGGAAGCCGGGCCA reverse read
444_1840_803_F3 has 0 matches
444_1840_980_F3 has 1 match with a score of 29:
2620828 GCACGAAAACGCCGCGTGGCTGGATGGT*CAAC*GTC 2620862 reference
||||||||||||||||||||||||||||*||||*|||
GCACGAAAACGCCGCGTGGCTGGATGGT*CAAC*GTC read
444_1840_1046_F3 has 1 match with a score of 32:
3673206 TT*GGTCAGGGTCTGGGCTTAGGCGGTGAATGGGGC 3673240 reference
||*|||||||||||||||||||||||||||||||||
TT*GGTCAGGGTCTGGGCTTAGGCGGTGAATGGGGC reverse read
444_1841_22_F3 has 0 matches
444_1841_213_F3 has 1 match with a score of 29:
1593797 CTTTG*AGCGCATTGGTCAGCGTGTAATCTCCTGCA 1593831 reference
|||||*|||||||| |||||||||||||||||||||
CTTTG*AGCGCATTAGTCAGCGTGTAATCTCCTGCA reverse read
The first alignment is a perfect match and scores 35 since the reads are all of length 35. The next alignment has two inferred color errors that each count is -3 (marked by * between residues), so the score is 35 - 2 x 3 = 29. Notice that the read is reported as the inferred sequence taking the color errors into account. The last alignment has one color error and one mismatch giving a score of 34 - 3 - 2 = 29, since the mismatch cost is 2.
Running the same reference assembly without allowing for color errors, the result is:
444_1840_767_F3 has 1 match with a score of 35:
1046535 GATACTCAATGCCGCCAAAGATGGAAGCCGGGCCA 1046569 reference
|||||||||||||||||||||||||||||||||||
GATACTCAATGCCGCCAAAGATGGAAGCCGGGCCA reverse read
444_1840_803_F3 has 0 matches
444_1840_980_F3 has 0 matches
444_1840_1046_F3 has 1 match with a score of 29:
3673206 TTGGTCAGGGTCTGGGCTTAGGCGGTGAATGGGGC 3673240 reference
|||||||||||||||||||||||||||||||||
AAGGTCAGGGTCTGGGCTTAGGCGGTGAATGGGGC reverse read
444_1841_22_F3 has 0 matches
444_1841_213_F3 has 0 matches
The first alignment is still a perfect match, whereas two of the other alignment now do not match since they have more than two errors. The last alignment now only scores 29 instead of 32, because two mismatches replaced the one color error above. This shows the power of including the possibility of color errors when aligning: many more matches are found.
The reference assembly program in CLC Genomics Workbench does not directly support alignment in color space only, but if such an alignment was carried out, sequence 444_1841_213_F3 would have three errors, since a nucleotide mismatch leads to two color space differences. The alignment would look like this:
444_1841_213_F3 has 1 match with a score of 26:
1593797 CTTTG*AGCGCATT*G*GTCAGCGTGTAATCTCCTGCA 1593831 reference
|||||*||||||||*|*|||||||||||||||||||||
CTTTG*AGCGCATT*G*GTCAGCGTGTAATCTCCTGCA reverse read
So, the optimal solution is to both allow nucleotide mismatches and
color errors in the same program when dealing with color space
data. This is the approach taken by the assembly program in CLC Genomics Workbench.
Note! If you set the color error cost as low as 1 while keeping the mismatch cost at 2 or above, a mismatch will instead be represented as two adjacent color errors.