Complex variant representations and VCF reference overlap
Allelic variants that overlap but do not cover the same range are called complex variants. Whenever two independently called variant sets are joined, there is a chance of getting complex variants. Complex variants comprise 1.4% of variants called by the CLC Fixed Ploidy Variant Detection tool. The popular GATK haplotype caller also encounters this phenomenon.
It is tricky to describe complex variants in VCF since they have to be written on different lines due to their position while they also need to be specified in the genotype field of each line without referring to the other lines. It may be possible to extend or split overlapping variants to match each other's position in order to comply with the VCF format, however that will lead to inaccuracies when assigning attributes, such as read count and coverage, to the altered variants.
GATK4 outputs complex variants with a genotype field that includes a reference allele for each overlapping allele, thereby also indicating that the variant is heterozygous. Since it means that two VCF lines will contradict each other, it can be argued that this representation is counter-intuitive. RTG tools' vcfeval provides an option called "-ref-overlap" to handle this representation. When interpreting complex variants represented this way, in case of conflict, non-reference alleles trump reference alleles.
To allow flexibility for communication with variant tracks, we provide the users with the following representation options for the VCF Export tool:
- The reference overlap representation as described above, where reference alleles are added to the genotype field of complex variants. We refer to this as the "Reference overlap" option. We also provide a version of the "Reference overlap" option with allele depth estimation.
- The legacy VCF export format (as available in previous versions of the software)
- The star allele format, based on the star allele introduced in VCF v4.2.
All of these complex variant representations can be handled by the VCF Import tool. A comparison of the options available is presented in figure 7.30:
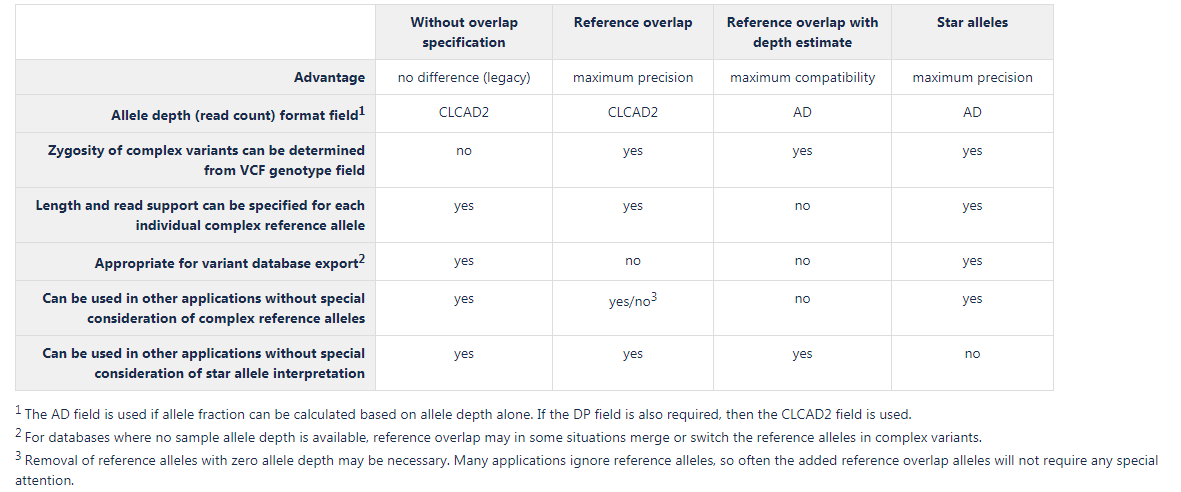
Figure 7.30: Main characteristics of the complex variant representations.
Without overlap specification
This is the representation used previously, where only variants that are present at the exact same ref positions are specified in the VCF genotype field. Variants that partially overlap do not affect the genotype field. Using this complex variant representation, two types of information are not available in the genotype field that is available for non-complex variants: zygosity of the variant, and the ploidy of the sample at the position.Suggested use cases: export of database variants without sample specific annotations (such as clinvar), where specification of sample haplotype structure is not necessary. Also use for applications tailored to handle this legacy format.
Reference overlap
This representation both allow specification of zygosity, ploidy, and phasing in the genotype field, as well as exact read support and length for complex reference alleles. At positions with complex alternate variants, a reference allele is specified in the VCF genotype field for each reference and alternate allele overlapping the position, these are termed reference overlap alleles. The allele depth is left at zero for reference overlap alleles, indicating that they are merely placeholders for overlapping alleles. The length and allele depth of complex reference alleles are specified separately, so the properties they have in the variant track are retained.Suggested use cases: this should be the general first choice, since it is an accurate representation of the variants, widely compatible with downstream applications
Reference overlap with depth estimate
This is the most compliant representation, where both the genotype and allele depth fields consider all alleles that overlap the position. In VCF files using the AD field for read count, it is common to be able to calculate allele frequency using the formula: frequency=AD/sum(AD), and that is also possible using this complex variant representation. The reference allele depth represents the combined read depth of overlapping alleles and reference alleles at the position, and is estimated as total read coverage (DP field) minus the combined allele depth of the ALT alleles at the position. This representation only specifies reference alleles together with alternate alleles. The main disadvantage of this representation is that it is not possible to specify exactly what the read support is for a complex reference allele, due to the fact that the reference allele depth is mixed with the overlapping allele depth. Complex reference alleles will get an average allele depth of the overlapping and reference alleles that are present at a position.Suggested use cases: export of variants for use in applications that cannot handle the more accurate "Reference overlap" representation
Star alleles
According to the VCF specification, star alleles are reserved for overlapping deletions, however some applications treat these in a way that is applicable to all types of overlapping variants. Since the overlapping deletion is defined in another VCF line, and it is unclear if the star allele signifies that the whole position is covered by the deletion, it is sometimes not appropriate to treat the star allele as an actual variant. The star allele can be interpreted merely as providing genotype information for the position, such as zygosity, ploidy, phasing and allele frequencies, whereas the actual overlapping variant will be dealt with at its start position where it is described in detail. This is the way the star allele is interpreted during VCF import in the CLC workbench. When using the star allele complex variant representation it is important to check if the variants are used in an application that handles the star alleles in a way similar to how the CLC workbench does, or if the star alleles are interpreted as actual deletion variants. In the latter case, another complex variant representation should be considered. This representation estimates the star allele depth, i.e. the number of reads supporting the overlapping alleles, to be the difference between the total read coverage and the combined allele depth of the variants at the position. Thus, the allele fraction can be calculated based on allele depth alone, and therefore the AD field is used for allele depth.Suggested use cases: This representation is accurate and does not require any special reference allele handling (no reference overlap). It should be used for all applications that handle star alleles as described above.
An example of export and import using the different complex variant representations is shown in figure 7.31:
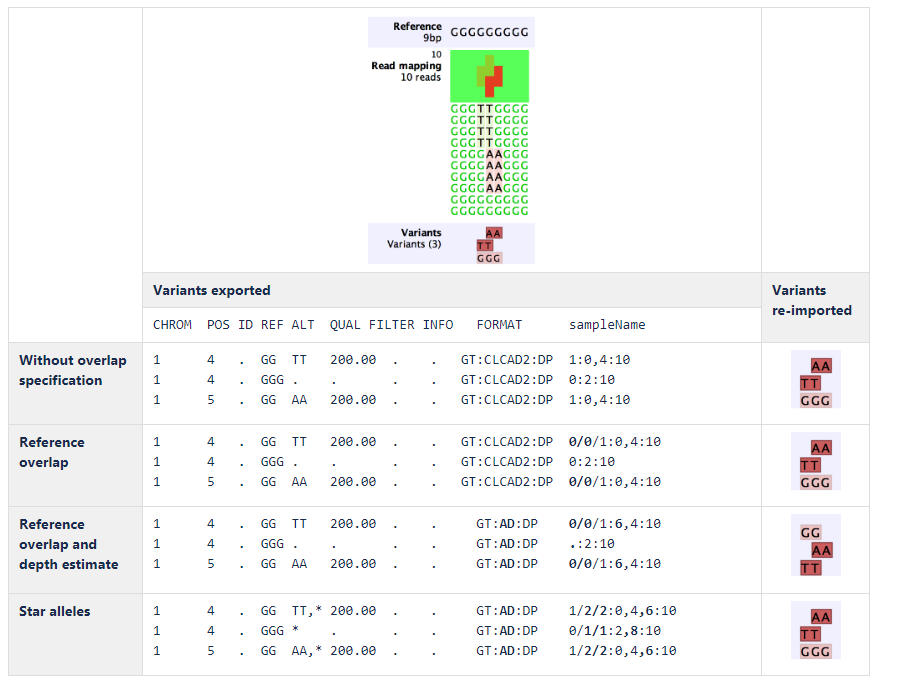
Figure 7.31: Example of export and import using the different complex variant representations.