Extract Reads
The Extract Reads tool extracts reads from read mappings, providing the extracted reads either in a track-based read mapping (reads track) or in a sequence list. Either all reads or a subset of reads can be extracted. Subsets can be specified based on location relative to a set of annotations and/or based on specified characteristics of the reads.
To launch Extract Reads, go to:
Toolbox | Utility Tools (![]() ) | Extract Reads (
) | Extract Reads (![]() )
)
Select a stand-alone read mapping or reads track as input (figure 35.3). To run the tool on multiple mappings, check the Batch checkbox.
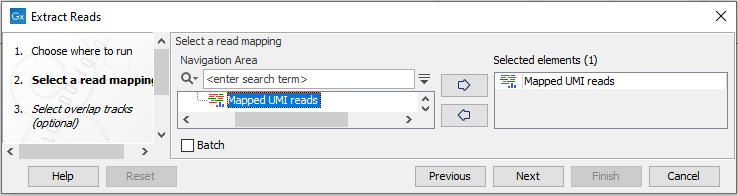
Figure 35.3: Select a read mapping as input. Here, a reads track has been selected.
The next wizard step (figure 35.4) is relevant if you wish to extract reads based on their location relative to a set of annotations or locations in an RNA-Seq statistical comparison track.
- Overlap tracks
- Overlap tracks When one or more tracks are provided, only reads that overlap one or more of the annotated regions in a specified way will be extracted. The reference genome for mappings and annotation tracks must be compatible to use this functionality. This means they contain the same number of chromosomes of corresponding lengths. This functionality can be used even if stand-alone read mappings were input, as long as the reference sequences of the mapping and overlap tracks are compatible.
- Type of overlap Specify how the reads must overlap the regions in the selected overlap tracks in order to be extracted.
- Any overlap. This will extract any reads that overlap regions in the overlap tracks (figure 35.5).
- Within region. Only include reads that are fully within the overlap track regions. That is, reads overlapping boundaries of the regions are not included (figure 35.5).
- Span region. Only extract reads that span the regions, i.e. have aligned residues on both sides of a region. For paired reads, fragments that span a region will be extracted. The option Only include matching read(s) of read pairs available in the next wizard step can be enabled to solely extract individual reads that span a region.
- No overlap. Extracts all reads except those overlapping a region in the overlap tracks (figure 35.5).
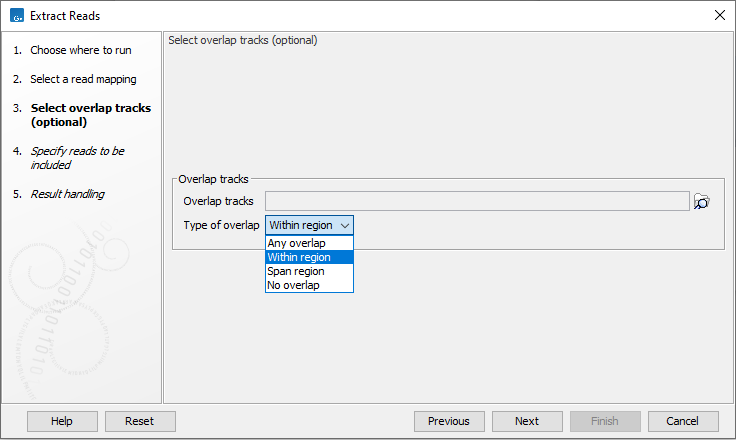
Figure 35.4: Specify overlap tracks to only extract reads mapped to particular areas of the reference genome.
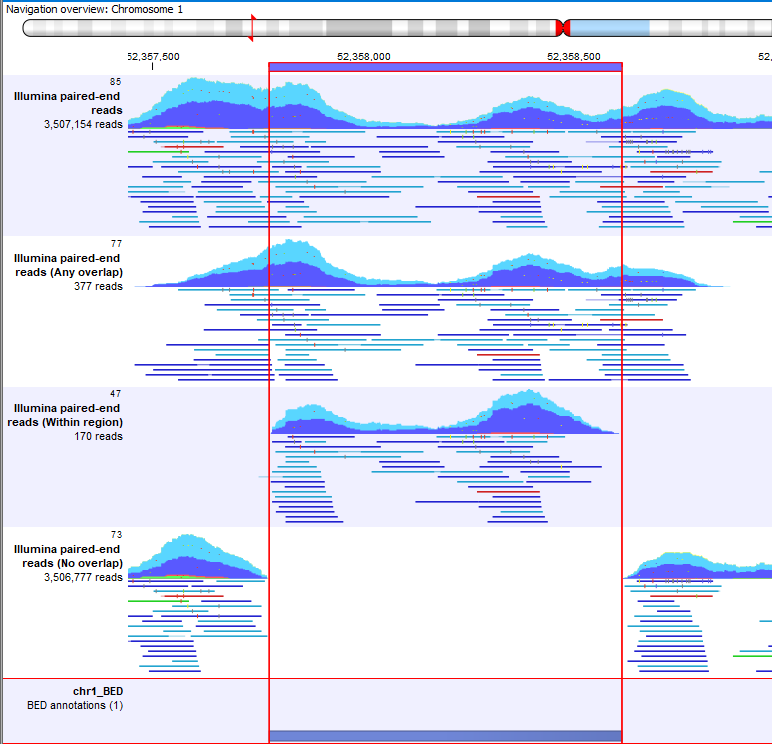
Figure 35.5: A track list including a full read mapping (top), read mappings where reads where extracted using the settings Any overlap, Within region or No overlap, and the overlap track defining the region reads were extracted against (bottom).
In the next wizard step, the nature of the reads to extract can be specified. All the options are enabled by default (figure 35.6).
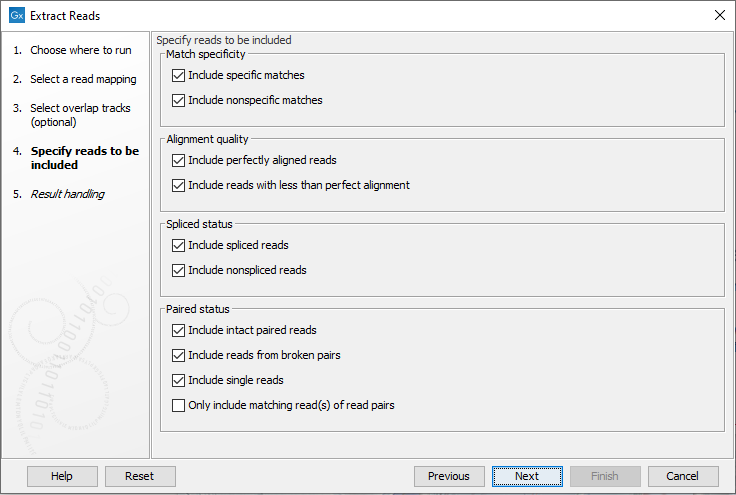
Figure 35.6: Options to include or exclude specific types of reads.
- Match specificity
- Include specific matches Reads that mapped best to just a single position of the reference genome.
- Include non-specific matches Reads that have multiple equally good alignments to the reference. These are the reads colored yellow by default in read mappings.
- Alignment quality
- Include perfectly aligned reads Reads where the full read is perfectly aligned to the reference genome (or consensus sequence for de novo assemblies). Reads that extend beyond the end of a contig are not considered perfectly aligned because part of the read does not match the reference.
- Include reads with less than perfect alignment Reads with mismatches, insertions or deletions, or with unaligned ends.
- Spliced status
- Include spliced reads Reads mapped across an intron.
- Include non spliced reads Reads not mapped across an intron.
- Paired status
- Include intact paired reads Paired reads mapped within the paired distance specified.
- Include reads from broken pairs Paired reads where only one of the reads mapped, either because only one read in the pair matched the reference, or because the distance or relative orientation of its mate was wrong.
- Include single reads Reads marked as single reads (as opposed to paired reads). Reads from broken pairs are not included in this category. Reads marked as single reads after trimming paired sequence lists are included in this category.
- Only include matching read(s) of read pairs If only the forward or reverse read of a read pair matches the criteria, then only include the matching read as a broken pair. For example if the forward read is inside the overlap region but the reverse read is not, then this option only includes the forward read as a broken read. When both forward and reverse reads are inside the overlap region then the full paired read is included. Note that some tools ignore broken reads by default.
In the last wizard step, the output type is selected. The options are to output reads tracks or sequence lists.
Reads in read mappings are colored according to their characteristics. The default color scheme is described in Coloring of mapped reads.