Stand-alone read mapping
Reads can be mapped to linear and circular chromosomes. Read mappings to circular genomes are visualized linearly as shown in figure 28.25.
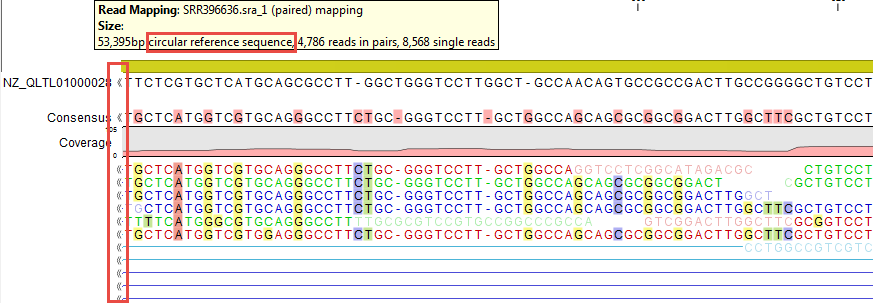
Figure 28.25: Mapping reads to a circular chromosome. Reads that are marked with double arrows at the ends are reads that map across the starting point of the sequence. The arrows indicate that the alignment continues at the other end of the reference sequence.
Reads that map across the starting point of the sequence are shown both at the start and end of the reference sequence. Such reads are marked with » at the end of the read to indicate that the alignment continues at the other end of the reference sequence.
Note that it is possible to select a portion of a read and access a right-click menu where you can Copy or Open in a New View the selected portion of the read: these option are available for the reference sequence at all compactness levels, and for individual reads at Low and Not compact levels.
If your read mapping or track shows the message 'Too much data for rendering' on a grey background, simply zoom in to see your reads in more detail. This occurs when there are too many reads to be displayed clearly. More specifically, where there are more than 500,000 reads displayed in a reads track, more than 200,000 reads displayed in a read mapping, or when the region being viewed in a read mapping is longer than 200,000 bases. Paired reads count as one in these cases.
Subsections