Subsample Sequence List
The Subsample Sequence List tool allows you to sample a nucleotide or protein sequence list by specifying either an absolute number or a percentage of the sequences to extract35.1.
Having very high numbers of sequences can be resource intensive without necessarily leading to higher quality results. In such cases subsampling a sequence list can be helpful. Examples of such situations are provided at the end of this section.
To run Subsample Sequence List:
Toolbox | Utility Tools (![]() ) | Sequence Lists (
) | Sequence Lists (![]() ) | Subsample Sequence List (
) | Subsample Sequence List (![]() )
)
In the first wizard step, select the sequence list to sample from (figure 35.16).
Figure 35.16: Select the sequence list to sample from.
In the next wizard step, configure the tool's settings (figure 35.17).
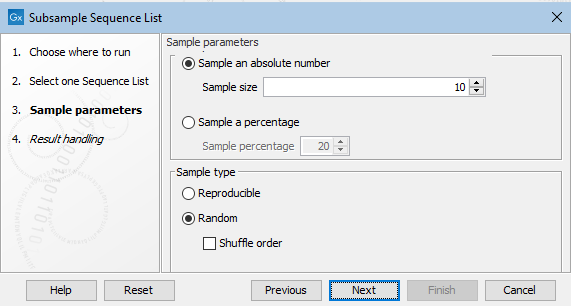
Figure 35.17: Details of the sample of sequences to extract are specfied at the Sample parameters step.
Sample size options
- Sample an absolute number
- Extract the number of sequences specified in the "Sample size" field from the sequence list provided as input.
- Sample a percentage
- Extract a percentage of the sequences from the sequence list provided as input. The percentage to extract is specified in the "Sample percentage" field.
Note: When working with paired-end reads, each pair is considered as 2 reads. Only complete pairs will be returned. For example, if a sequence list contains 3 paired reads (6 sequences), and a sample of 50% was requested, then the 2 sequences of a single pair would be returned. If an odd number of reads is requested, an even number would be returned. For example, if 3 reads were requested, 2 would be returned. By contrast, if 6 single end reads were provided as input, 3 reads would be returned in both cases.
Sample type options
- Reproducible
- Return the same set of sequences each time the tool is run with the same input and sample size options specified.
- Random
- Return a different subset of sequences each time the tool is run with the same input and sample size options specified.
- Shuffle
- When unchecked, sequences returned using the Random option will in the same order as they appeared in the original sequence list. When checked, the order of the sequences in the output is shuffled.
Examples of situations to subsample sequence lists
Mapping reads with a very deep coverage can lead to slow response times, especially when the mappings will be investigated in detail. For example, the speed of edit operations in stand-alone mappings after running a tool like Map Reads to Contigs can be slowed down by excessive read depth. Subsampling very large sequence lists before mapping can thus lead to better performance, often without loss of important information, depending on the situation.
For de novo assembly, very high coverage in a location increases the probability that the information in that region is interpreted as being associated with sequencing errors. Reducing the coverage to a maximum average of 100x can thus be a good idea for this application. The following calculation can be used for this purpose:
- Multiply the estimated size of the genome you intend to assemble by 100 to give the total number of bases to use as input for the de novo assembly.
- Divide this total number of bases by the average length of the reads.
- Specify the result of this calculation as the absolute number of reads to sample from the sequence list.
See the De novo sequencing chapter for further details about running a de novo assembly and mapping reads to contigs.
Footnotes
- ... extract35.1
- Prior to CLC Genomics Workbench 22.0, this tool was called Sample Reads