How to run the InDels and Structural Variants tool
To start the structural variant detection:
Toolbox | Resequencing (![]() ) | InDels and Structural Variants tool (
) | InDels and Structural Variants tool (![]() )
)
This will open up a dialog. Select the read mapping of interest as shown in figure 26.18 and click on the button labeled Next.
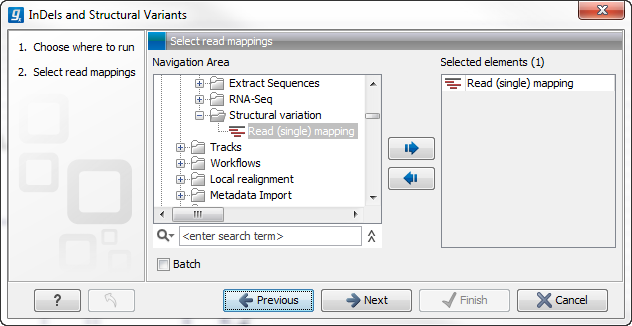
Figure 26.18: Select the read mapping of interest.
Specify the settings shown below and in figure 26.19. For further details about these settings, please see Creating Left- and Right breakpoint signatures.
- Significance of unaligned end breakpoints
- The P-value threshold The threshold for calling significance in the binominal distribution of unaligned end reads.
- Maximum number of mismatches When examining positions for excess of unaligned ends, only reads mapped with this or fewer mismatches will be considered.
- Filter variants
- Filter variants Allows the user to filter out structural variants that derived from breakpoints that are supported by few reads.
- Minimum number of reads The minimum number of reads supporting the breakpoints used to infer the variant before this variant will be reported.
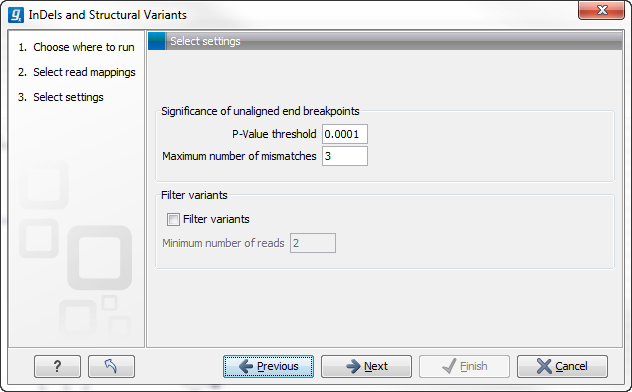
Figure 26.19: Select the relevant settings.
Clicking on the button labeled Next opens up the "Results handling" dialog (figure 26.20) with the following output options:
- Create report Creates a report that summarizes information about the observed breakpoints and variants.
- Create breakpoints Creates a track showing the detected breakpoints.
- Create InDel variants Creates a variant track containing the detected InDels that fulfill the requirements for being 'variants'. (These include the detected insertions for which the allele sequence is inferred, but not those for which it is not, or only partly, known. Also, only deletions of six up to 200 bp are included in the variant track. See Variant tracks for a definition of the requirements for 'variants'). Note that insertions and deletions that are not included in the InDel track, will be present in the 'Structural variants track').
- Create structural variations Creates a track showing the detected structural variants.
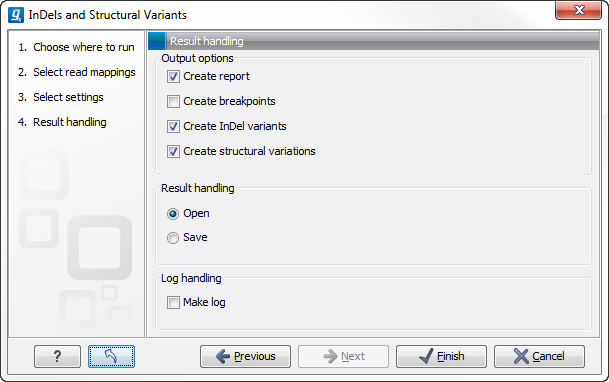
Figure 26.20: Select output formats.