Defining reference genome and mapping settings
You are now presented with the dialog shown in figure 27.4.
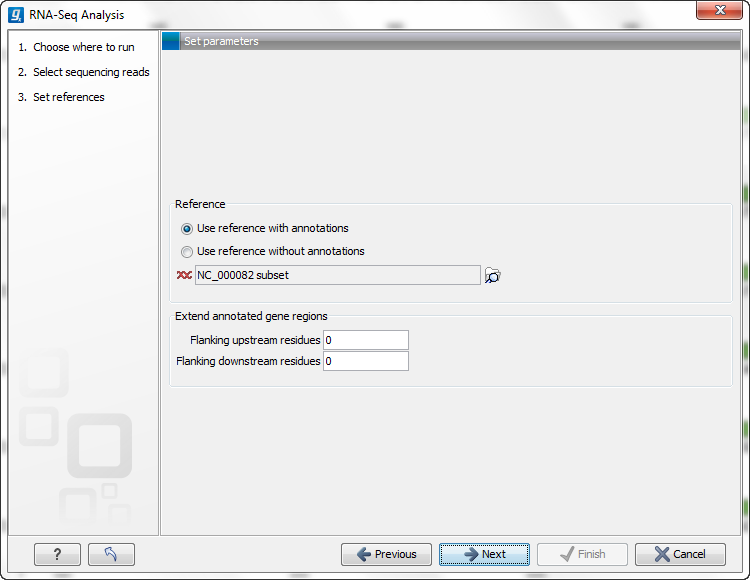
Figure 27.4: Defining a reference genome for RNA-Seq.
At the top, there are two options concerning how the reference sequences are annotated:
- Use reference with annotations. Typically, this option is chosen when you have an annotated genome sequence. Choosing this option means that gene and mRNA annotations on the sequence will be used if you choose the option Eukarotes in the next window. If you choose the option Prokaryotes in the next window, the annotations of type gene only are used. See Finding the right reference sequence for RNA-Seq.
- Use reference without annotations. This option is suitable for situations like mapping back reads to un-annotated EST consensus sequences. The reference in this case is a list of sequences. A common situation is for a multi-fasta file to be imported into the Workbench to be used for this purpose. Each sequence in the list will be treated as a "gene" (or "transcript"). Note that the Workbench uses prokaryote settings here. This means that it does not look for new exons (see Exon discovery) and it assumes that the sequences have no introns).
Just below these two options, you click to select the reference sequences.
Next, you can choose to extend the region around the gene to include more of the genomic sequence by changing the value in Flanking upstream/downstream residues. This also means that you are able to look for new exons before or after the known exons (see Exon discovery).
When the reference has been defined, click Next and you are presented with the dialog shown in figure 27.5.
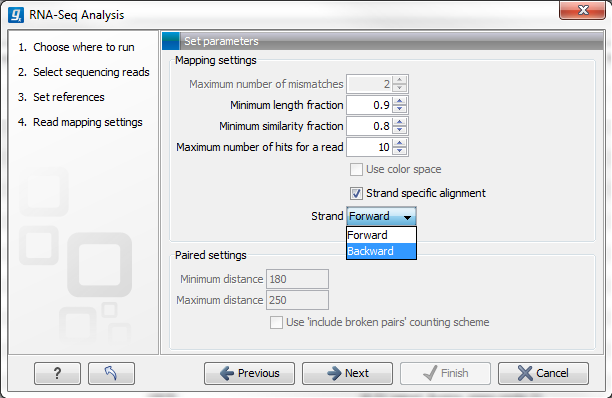
Figure 27.5: Defining mapping parameters for RNA-Seq.
Different mapping algorithms are applied when mapping the reads in sequence lists containing only short reads (those under 56bp in length) and when mapping reads in sequence lists containing one or more reads that are 56bp or longer. The mapping algorithm used is applied to all reads in a given sequence list. Different algorithms are not used for particular reads within a given sequence list.
Accordingly, the mapping parameters made available to edit via the Wizard depend on the read lengths in the sequence lists. If at least one sequence list containing only short sequences (those under 56bp in length) was entered, then the "Maximum number of mismatches" setting will be available to edit. If at least one sequence list of reads containing at least one read 56bp or longer was entered, then the "Minimum length fraction" and "Minimum similarity fraction" settings will be available. If you have entered multiple sequence lists, some lists containing only short reads and some lists containing at least one or more longer reads, then all the mapping parameter settings will be made available for editing. The "Maximum number of mismatches" setting will be used only for the mapping of the lists containing all short reads. The "Minimum length fraction" and "Minimum similarity fraction" settings will be used only for the mapping of all entries in sequence lists where one or more of the reads is 56bp or longer.
The mapping parameters are:
- Maximum number of mismatches. This parameter is available if you have selected at least one sequence list containing only short reads (shorter than 56 nucleotides, except in the case of color space data, which are always treated as long reads). This is the maximum number of mismatches to be allowed. Maximum value is 3, except for color space where it is 2.
- Minimum length fraction. This parameter is available when at least one sequence list entered contains sequence(s) 56bp or longer. It specifies how much of a read must match to the reference to the level of similarity specified in the last parameter for this read to be mapped. The default is 0.9 which means that at least 90 % of the bases need to align to the reference.
- Minimum similarity fraction. This parameter is available when at least one sequence list entered contains sequence(s) 56bp or longer. It specifies how similar the matching part of the read should be to the reference, for that read to be mapped. When using the default setting at 0.8 and the default setting for the length fraction, it means that 90 % of the read should align with 80 % similarity in order to include the read.
- Maximum number of hits for a read.
A read that matches to more distinct places in the references than the 'Maximum number of hits for a read' specified will not be mapped (the notion of distinct places is elaborated below). If a read matches to multiple distinct places, but below the specified maximum number, it will be randomly assigned to one of these places. The random distribution is done proportionally to the number of unique matches that the genes to which it matches have, normalized by the exon length (to ensure that genes with no unique matches have a chance of having multi-matches assigned to them, 1 will be used instead of 0, for their count of unique matches). This means that if there are 10 reads that match two different genes with equal exon length, the 10 reads will be distributed according to the number of unique matches for these two genes. The gene that has the highest number of unique matches will thus get a greater proportion of the 10 reads.
Places are distinct in the references if they are not identical once they have been transferred back to the gene sequences. To exemplify, consider a gene with 10 transcripts and 11 exons, where all transcripts have exon 1, and each of the 10 transcripts have only one of the exons 2 to 11. Exon 1 will be represented 11 times in the references (once for the gene region and once for each of the 10 transcripts). Reads that match to exon 1 will thus match to 11 of the extracted references. However, when transferring the mappings back to the gene it becomes evident that the 11 match places are not distinct but in fact identical. In this case the read will not be discarded for exceeding the maximum number of hits limit, but will be mapped. In the RNA-seq action this is algorithmically done by allowing the assembler to return matches that hit in the 'maximum number of hits for a read' plus 'the maximum number of transcripts' that the genes have in the specified references. The algorithm post-processes the returned matches to identify the number of distinct matches and only discards a read if this number is above the specified limit. Similarly, when a multi-match read is randomly assigned to one of it's match places, each distinct place is considered only once.
- Strand-specific alignment. When this option is checked, the user can specify whether the reads should be attempted mapped only in their forward (or reverse) orientation. This will typically be appropriate when a strand specific protocol for read generation has been used. It allows assignment of the reads to the right gene in cases where overlapping genes are located on different strands. Without the strand-specific protocol, this would not be possible (see [Parkhomchuk et al., 2009]). Also, applying the 'strand specific' 'reverse' option in an RNA-seq run, to reads that did not map in a 'strand specific' 'forward' RNA-seq run, will allow the user to assess the degree of antisense transcription.
Subsections