Pattern Discovery
With CLC Main Workbench you can perform pattern discovery on both DNA and protein sequences. Advanced hidden Markov models can help to identify unknown sequence patterns across single or even multiple sequences.
In order to search for unknown patterns:
Tools | General Sequence Analysis (![]() )| Pattern Discovery (
)| Pattern Discovery (![]() )
)
Choose one or more sequence(s) or sequence list(s). You can perform the analysis on several DNA or several protein sequences at a time. If the analysis is performed on several sequences at a time the method will search for patterns which is common between all the sequences. Annotations will be added to all the sequences and a view is opened for each sequence.
Click Next to adjust parameters (see figure 19.19).
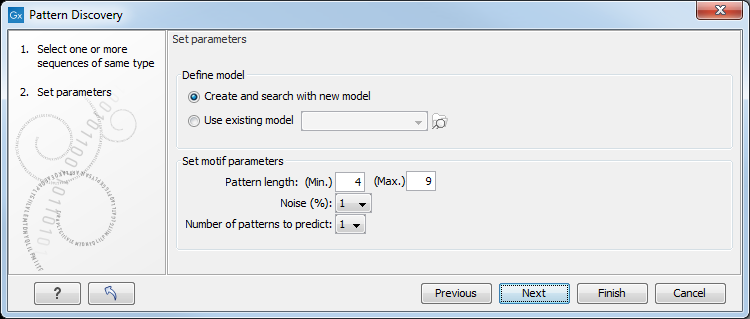
Figure 19.19: Setting parameters for the pattern discovery. See text for details.
In order to search unknown sequences with an already existing model:
Select to use an already existing model which is seen in figure 19.19. Models are represented with the following icon in the Navigation Area (![]() ).
).
Pattern discovery search parameters
Various parameters can be set prior to the pattern discovery. The parameters are listed below and a screenshot of the parameter settings can be seen in figure 19.19.
- Create and search with new model. This will create a new HMM model based on the selected sequences. The found model will be opened after the run and presented in a table view. It can be saved and used later if desired.
- Use existing model. It is possible to use already created models to search for the same pattern in new sequences.
- Minimum pattern length. Here, the minimum length of patterns to search for, can be specified.
- Maximum pattern length. Here, the maximum length of patterns to search for, can be specified.
- Noise (%). Specify noise-level of the model. This parameter has influence on the level of degeneracy of patterns in the sequence(s). The noise parameter can be 1,2,5 or 10 percent.
- Number of different kinds of patterns to predict. Number of iterations the algorithm goes through. After the first iteration, we force predicted pattern-positions in the first run to be member of the background: In that way, the algorithm finds new patterns in the second iteration. Patterns marked 'Pattern1' have the highest confidence. The maximal iterations to go through is 3.
- Include background distribution. For protein sequences it is possible to include information on the background distribution of amino acids from a range of organisms.
The patterns found are added as annotations on the original sequence (see figure 19.20).

Figure 19.20: Sequence view displaying two discovered patterns.
Pattern search output
If the analysis is performed on several sequences at a time the method will search for patterns in the sequences and open a new view for each of the sequences, in which a pattern was discovered. Each novel pattern will be represented as an annotation of the type Region. More information on each found pattern is available through the tool-tip, including detailed information on the position of the pattern and quality scores.
It is also possible to get a tabular view of all found patterns in one combined table. Then each found pattern will be represented with various information on obtained scores, quality of the pattern and position in the sequence.
A table view of emission values of the actual used HMM model is presented in a table view. This model can be saved and used to search for a similar pattern in new or unknown sequences.