BLAST at NCBI
Use BLAST at NCBI to launch a BLAST search using query sequences you select in your Navigation Area. The search is run on the NCBI's BLAST servers. When the results are ready, they are automatically downloaded, ready to view in your CLC Main Workbench.
When a large number of query sequences are selected, they will be split into subsets, with each subset sent individually to the NCBI. This helps avoid exceeding any internal limits the NCBI places on the number of sequences in a single submission. The size of the subset created by CLC software depends both on the number and size of the sequences.
To start BLAST at NCBI, go to:
Tools | BLAST (![]() )| BLAST at NCBI (
)| BLAST at NCBI (![]() )
)
A keyboard shortcut is available: Ctrl+Shift+B (Windows) or ![]() +Shift+B (mac).
+Shift+B (mac).
When the query involves just a region of a single sequence, launching the tool directly from an open view of the sequence or sequence list may be preferable:
select the region of interest in the sequence |
right-click on the selected region | choose BLAST Selection Against
NCBI (![]() )
)
Specify the query sequences
After launching BLAST at NCBI from the Tools menu, one or more sequences or sequence lists of the same type, DNA or protein, are selected to search with (figure 27.6).
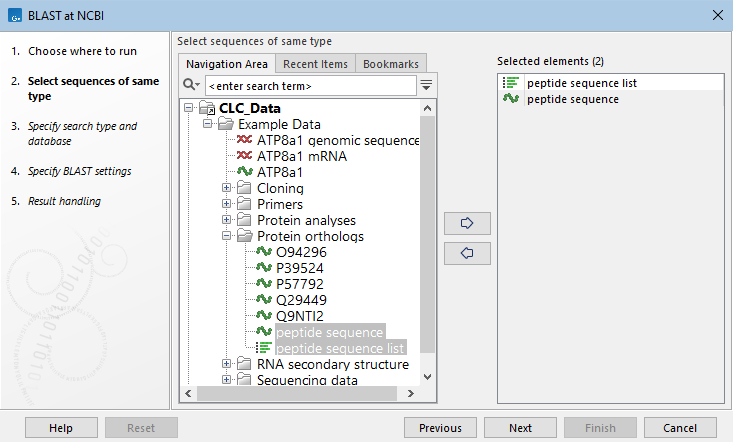
Figure 27.6: Specify one or more sequences or sequence lists to search with.
Specify the search type and database to search
In the next wizard step, the type of BLAST search and the database to search are specified (figure 27.7). Only databases relevant to the selected search type will be listed.
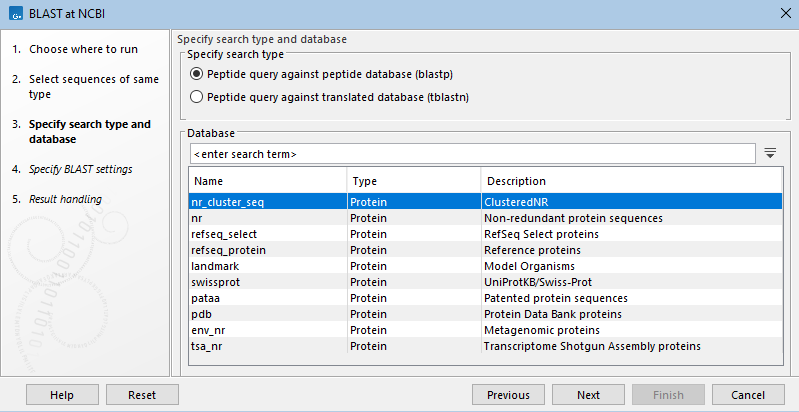
Figure 27.7: Specify the type of search to run and the database to search. Here, blastp has been selected, so available peptide databases are listed.
BLAST search types for nucleotide query sequences:
- Nucleotide query against nucleotide database When this type of search is specified, an option is available in the next wizard step to specify the type of blastn search to run. Megablast, which is designed to find very similar sequences (e.g. >95% similarity), is the default. Further details about this are below.
- Translated query against peptide database (blastx) DNA query sequences, translated in six frames, are used to search the selected peptide database. The genetic code to use to translate the query sequences is specified in the next wizard step.
- Translated query against a translated database (tblastx) DNA query sequences, translated in six frames, are used to search the selected nucleotide database, the entries of which are also translated in six frames. The genetic code to use to translate the query and the database are specified individually in the next wizard step. This type of search is computationally intensive.
BLAST search types for peptide query sequences:
- Peptide query against peptide database (blastp)
- Peptide query against translated DNA database (tblastn) Peptide query sequences are used to search a nucleotide database, which is translated in six frames using the genetic code specified in the following wizard step. This type of search is computationally intensive.
Refine the BLAST search options
In the following wizard step, search settings can be refined. The options available depend on the type of search being run. Available options are described briefly below. Most are described in more detail at https://blast.ncbi.nlm.nih.gov/doc/blast-topics/blastsearchparams.html.
- Optimization This section is shown for searches against a nucleotide database. When searching for somewhat similar sequences (blastn), word size, gap costs and match scoring can be adjusted. The default is to optimize for finding highly similar sequences (megablast). Using megablast, word size, gap costs and match scoring settings are locked.(See figure 27.8.)

Figure 27.8: When searching a nucleotide database with a nucleotide query sequence, the megablast task, designed to search for highly similar sequences, is the default optimization option. When this option is selected, word size, gap costs and match scoring are locked. - Choose genetic code A drop-down list of genetic codes is available when DNA query sequences will be translated before searching (blastx, tblastx) and when the translation of a DNA database will be searched (tblastn, tblastx).(See figure 27.9.)
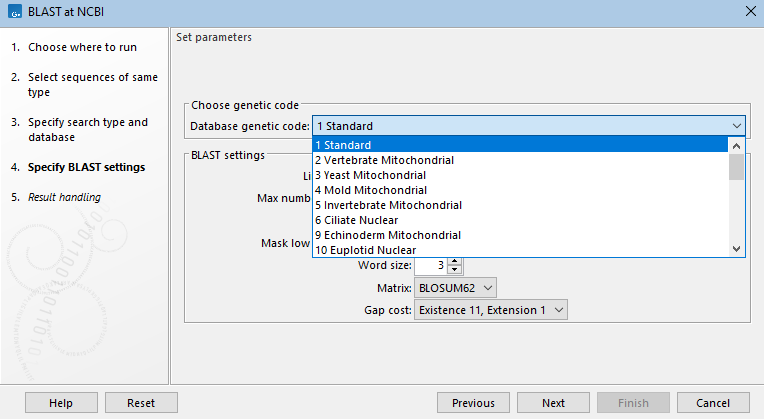
Figure 27.9: For tblastn searches, a nucleotide database is selected, but the search is run against a translation of that database in 6 frames. The genetic code to use for the translation is selected from a drop-down list. - Limit by Entrez query BLAST searches at the NCBI can be limited to a subset of the selected database based on an Entrez query entered in this field. Some commonly used Entrez queries are available in the drop down menu, but any terms normally allowed in an Entrez search can be entered in this field. More information about Entrez queries can be found at https://www.ncbi.nlm.nih.gov/books/NBK3837/#EntrezHelp.Entrez_Searching_Options. (See figure 27.10)
Figure 27.10: A drop-down list of common entrez limits is available, but custom limits can be entered into the field, as has been done here, where the BLAST search will be limited to searching against database entries recorded as being from the organism Clostridium botulinum. - Max number of hit sequences The maximum number of database sequence matches to include in the BLAST report.
- Expect Matches with an Expect value (E-value) greater than the value in this field will not reported.
Values lower than 1 can be entered as decimals, or in scientific notiation. For example, 0.001, 1e-3 and 10e-4 would be equivalent and acceptable values.
An E-value is a statistical measure representing the number of hits of a given quality, or better, that you would expect to see purely by chance in a database of the same size as the one being searched. I.e. The higher the E-value, the higher the chance that the match is not due to biological similarity with the query sequence. Details of how E-values are calculated can be found at the NCBI: https://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html.
- Mask low complexity regions Mask segments of the query sequence with low compositional complexity. This can reduce the number of hits reported that are statistically significant but biologically uninteresting (e.g. hits against common acidic-, basic- or proline-rich regions).
- Word Size BLAST starts off by finding word-sized matches between the query and database sequences and then initiating extensions from these matches. Thus the sensitivity and speed of a blastn search can be tuned by increasing or decreasing the word size.
For nucleotide-nucleotide searches an exact match of a word is required before an extension is initiated. The word size for megablast is locked at 28, but the word size for blastn searches can be adjusted.
For searches against peptide databases (including translated nucleotide), non-exact word matches are taken into account based upon the similarity between words. Word sizes of 2 or 3 are common for such searches.
- Match/mismatch When searching against a nucleotide database, scoring includes assigning a positive value when a base in the query matches a base in the database sequence (match) and a negative value when the bases do not match (mismatch). Match/mismatch scores are locked for megablast.
- Matrix When searching against peptide databases (including translated nucleotide databases), scoring makes use of an amino acid substitution matrix. A selection are available from the drop-down list, with BLOSUM62 set as the default. See https://www.ncbi.nlm.nih.gov/books/NBK279684/#appendices.BLAST_Substitution_Matrices for further details.
- Gap Cost The cost to open a gap (existence) and the cost to extend a gap (extension) in the alignment between the query and target sequences. See https://www.ncbi.nlm.nih.gov/books/NBK279684/#appendices.BLASTN_rewardpenalty_values for further information about gap costs.
Click on Finish to launch the analysis.