Find open reading frames
Find Open Reading Frames identifies open reading frames (ORFs) in sequences, and can be used as a rudimentary gene finder.
During translation of a transcript, protein is generated from the first start codon to the stop codon, internal start codons are translated to their respective amino acids. Find Open Reading Frames correspondingly always reports ORFs using the first possible start codon and ignores internal start codons.
Identified ORFs are shown as annotations on the sequence. Different genetic codes are available, but it is also possible to manually specify start codons.
In one analysis, Find Open Reading Frames can process a maximum of 100,000 sequences or 50 million base pairs. Sequences may be provided to the tool as individual sequences or as sequence lists.
To run Find Open Reading Frames, go to:
Tools | Nucleotide Analysis (![]() )| Find Open Reading Frames (
)| Find Open Reading Frames (![]() )
)
This opens the dialog displayed in figure 20.7
Figure 20.7: Select a sequence or a sequence list as input.
Use the arrows to add or remove sequences or sequence lists from the selected elements list.
Next, specify which parameters should be used (figure 20.8)
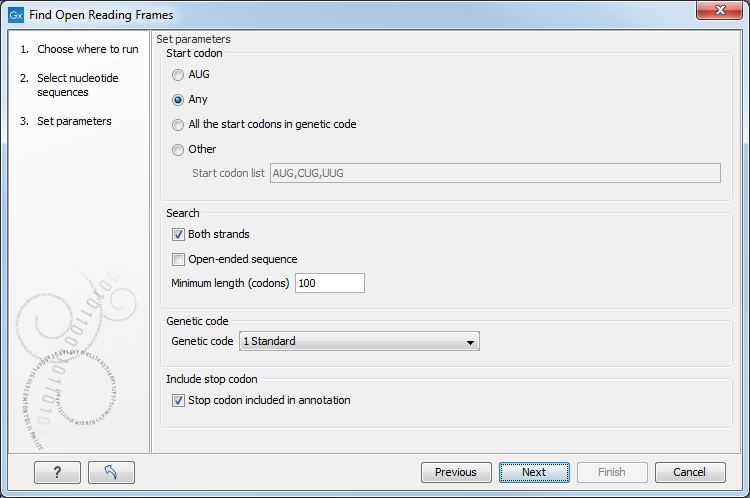
Figure 20.8: Configure the options for finding open reading frames. Hover the mouse cursor over a genetic code option to reveal a tooltip containing the relevant translation table.
- Start codon
- AUG Most commonly used start codon. When selected, only AUG (or ATG) codons are used as start codons.
- Any Any codon can be used as the start codon. For identification of the open reading frames, the first possible codon in the same reading frame as the stop codon is used as the start codon.
- All the start codons in genetic code Select to use the start codons that are specific to the genetic code specified under Genetic code.
- Other Identifies open reading frames that start with one of the codons provided in the start codon list.
- Both strands Find reading frames on both strands.
- Open-ended sequence Allow ORFs to extend up to the sequence start or end not considering the sequence context. This can be relevant when only a fragment of a sequence is analyzed, and there may be up- or downstream start and stop codons that are not included in the sequence. When predicting the open reading frames, stop codons are always used, but a given start codon is only used if it is the first one after the last stop codon. Start codons that are not preceded by a stop codon are ignored, because there may be another start codon upstream that is not included in the sequence.
- Minimum length (codons) The minimum number of codons that must be present for an open reading frame to be reported.
- Genetic code Specify the genetic code to use. Hover the mouse cursor over an item in this list to reveal a tooltip containing the relevant translation table (figure 20.8). The translation tables are sourced from the NCBI (https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi).
- Stop codon included in annotation Include the stop codon in the open reading frame annotations on the sequences.
Using open reading frames to find genes is a fairly simple approach which is likely to predict genes which are not real. Setting a relatively high minimum length of the ORFs will reduce the number of false positive predictions, but at the same time short genes may be missed (see figure 20.9).
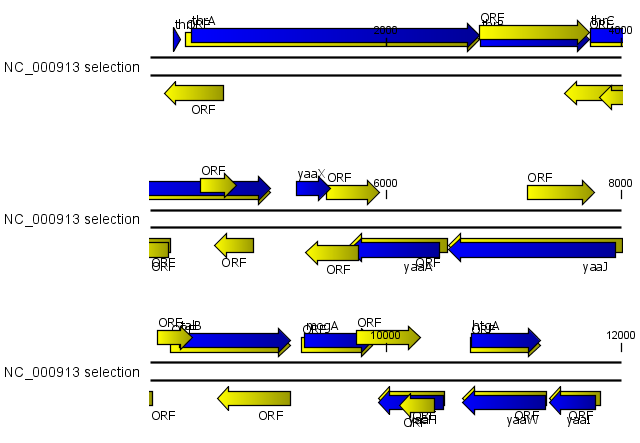
Figure 20.9: The first 12,000 positions of the E. coli sequence NC_000913 downloaded from GenBank. The blue (dark) annotations are the genes while the yellow (brighter) annotations are the ORFs with a length of at least 100 amino acids. On the positive strand around position 11,000, a gene starts before the ORF. This is due to the use of the standard genetic code rather than the bacterial code. This particular gene starts with CTG, which is a start codon in bacteria. Two short genes are entirely missing, while a handful of open reading frames do not correspond to any of the annotated genes.
Click on Finish to launch the analysis.
Finding open reading frames is often a good first step in annotating sequences such as cloning vectors or bacterial genomes. For eukaryotic genes, ORF determination may not always be very helpful since the intron/exon structure is not part of the algorithm.