Immune Repertoire Analysis from Reads (10xV(D)J)
The workflow Immune Repertoire Analysis from Reads (10xV(D)J) takes 10xV(D)J reads as input and starts by annotating them with cell barcode and UMI, followed by clonotype identification and filtering. The workflow uses iterate functionality and allows for a combined analysis of multiple samples to produce a single, multi-sample, filtered TCR Cell Clonotypes (The workflow can be found here:
Template Workflows | Single Cell Workflows (![]() ) | From Reads (
) | From Reads (![]() ) | Immune Repertoire Analysis from Reads (10xV(D)J) (
) | Immune Repertoire Analysis from Reads (10xV(D)J) (![]() )
)
If you are connected to a CLC Server via the CLC Single Cell Analysis Module, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
You can choose either one or more Sequence lists or Select files for on-the-fly import and select FASTQ files for importing.
The workflow is configured for 10x Chromium Single Cell V(D)J reads and only clonotype filtering is customizable. Adjustments can be made in a workflow copy, see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Creating_editing_workflows.html.
The workflow can be run using Single Cell hg38 (Ensembl) or Single Cell Mouse (Ensembl) reference data sets (see Reference data management).
|
Note: Reference data elements cannot be configured during workflow execution. If other elements than those provided in the default reference data sets are needed, a custom reference data set can be used, see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Reference_Data_Sets_defining_Custom_Sets.html. Reference V, D, J and C gene segments for other species or for B cells can be imported using Import Immune Reference Segments. |
The workflow allows the analysis of multiple samples and you can specify metadata during workflow execution for configuring which inputs belong to which sample. When there is only one library per sample, metadata is not necessary and "Use organization of input data" can be used. For more details on configuring workflow execution with metadata, see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Running_workflows_in_batch_mode.html. Make sure to inspect the batch overview to check that the analysis will be performed correctly.
Examples for how to use metadata for workflow execution can be found in Configuring the batch units for Expression Analysis from Reads.
The Filter Cell Clonotypes (![]() ) tool in the workflow is configured with most parameters locked, see 19.19. There are two open options. "Chains to retain" allows selecting the chains that are expected to be found, and removes noise from the results. "Multiple clonotypes" allows different handling of barcodes with more than one clonotype. The default option Retain primary retains only the clonotype with the highest number of reads. See Filter Cell Clonotypes for details.
) tool in the workflow is configured with most parameters locked, see 19.19. There are two open options. "Chains to retain" allows selecting the chains that are expected to be found, and removes noise from the results. "Multiple clonotypes" allows different handling of barcodes with more than one clonotype. The default option Retain primary retains only the clonotype with the highest number of reads. See Filter Cell Clonotypes for details.
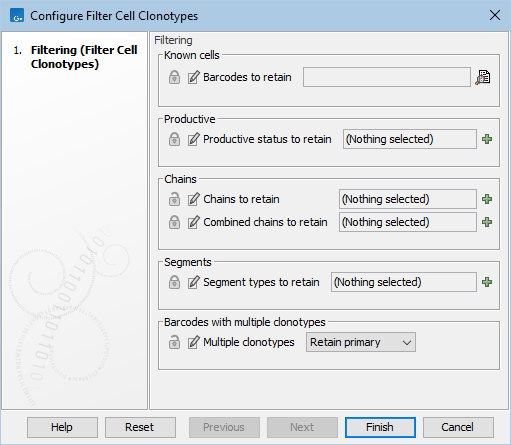
Figure 19.19: Workflow settings of Filter Cell Clonotypes.
Subsections