Configuring the batch units for Expression Analysis from Reads
When there is only library per sample, metadata is not necessary for workflow execution. Let us consider the FASTQ files shown in figure 19.1.

Figure 19.1: Example of ten FASTQ files for paired reads, originating from multiple lanes and three libraries.
The files can be automatically imported during workflow execution by choosing "Select files for on-the-fly import", selecting the Illumina importer and enabling "Paired reads" and "Join reads from different lanes". Selecting "Use organization of input data" when defining the batch units will lead to the input files being grouped in three libraries, as shown in figure 19.2. Note that if any of the samples has more than 1 billion paired reads, the metadata approach described below should be used instead.
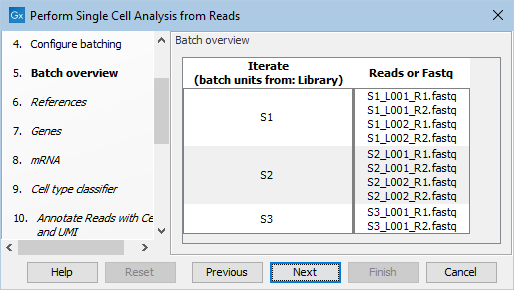
Figure 19.2: Batch overview when importing the FASTQ files and choosing "Use organization of input data".
Now let us consider the metadata shown in figure 19.3.
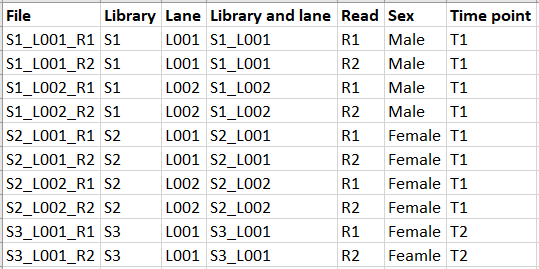
Figure 19.3: Example of metadata for the files from figure 19.1.
Metadata can be imported directly from an Excel or txt file during workflow execution and for this example, the "Library" metadata column should be used for defining the batch units (see figure 19.4).
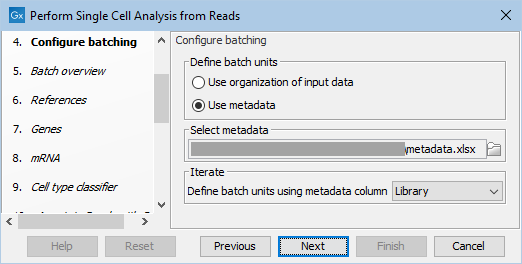
Figure 19.4: Configuring the workflow execution using metadata.
The workflow will automatically associate the input files with the correct rows in the metadata based on the first column and a batch overview similar to that in figure 19.2 will be shown. The additional metadata columns will be converted to cell annotations.
Note that in this workflow it is not possible to freely choose the batch units. Instead, each batch must correspond to one sample. The reason for the restriction is that each read is linked to a cell by the cell barcode. Batching by sample is required in order to inform the workflow that if the same cell barcode is present in multiple files, it is because it is the same cell.
Failing to batch by sample will likely lead to misleading results. For example, in figure 19.4 it would be necessary to batch by library. If we batched by "Time point", then two cells with the same barcode at time point T1 would be treated as being identical, even if one came from sample S1 and the other from sample S2. If, on the other hand, we batched by "Library and Lane", then a cell from sample S1 that was sequenced on both lanes would be split up into two cells - one for each lane.
The workflow combines all inputs to produce just one matrix. All metadata, including "Sex" and "Time point" in the provided example, will be available in the output cell annotations.
The FASTQ and metadata files can also be imported manually and used for the workflow execution.